
Chapter 7
Liquid Crystal-Functionalized Nano- and Microfibers Produced by Electrospinning
7.1 Introduction: Why Electrospinning With Liquid Crystals?
Although electrospinning is actually a rather old invention, it is currently attracting rapidly increasing attention throughout the international research community [1–8]. Among the reasons is the potential of the technique for producing extremely thin polymer fibers, which can easily be functionalized by inorganic as well as organic non-polymeric additives. By including liquid crystals inside the fibers, or by spinning the fibers from liquid crystalline polymers, a number of interesting phenomena arise [9–15], inspiring new directions of academic research and holding potential for innovative applications. The extreme stretching during the spinning process aligns a main-chain liquid crystalline polymer very well along the fiber [15], and electrospinning a lyotropic cellulose-based chiral nematic phase results in helical fibers [14]. If a low molar mass liquid crystal is enclosed inside the polymer fiber the strong encapsulation in a cylindrical space with diameter in the nano-to micrometer range can have dramatic effects, e.g., expanding the liquid crystal phase sequence [12], inducing unusual structures of the director field or restricting normally continuously varying parameters to quantized values [10]. Compared to other approaches of studying liquid crystal confinement effects, electrospinning has the attractive property that the mesogenic material is incorporated at the time of producing the fiber; thus no time-consuming filling procedure is required. On the other hand the various types of unique properties of different liquid crystal phases—strong response function to applied fields or temperature changes, birefringence, photonic band gap, spontaneous polarization, etc.—can all be transferred to the polymer fiber, giving it a responsiveness and functionality that regular polymer fibers do not exhibit. This is wherein the main potential for applications lies, relevant in particular for the field of wearable electronics and smart textiles.
Liquid crystal electrospinning is a very young research field and it is presently studied by only a handful of groups around the world. Considering the numerous opportunities for curiosity-based as well as applied research and the simplicity and low investment costs of the experimental equipment a strong development in the near future can be expected. This chapter aims to introduce the concept, describe the basic practical requirements, and give some examples of current achievements as well as an outlook to where the field is heading, discussing also the application potential. It is the hope of the author that this will inspire new players to enter the field, taking it into new directions and initiating further original thrusts in the field. It is still largely unexplored and thus offers many attractive opportunities for innovative research.
7.2 What is Electrospinning?
Electrospinning provides a convenient means of producing exceptionally thin fibers from a variety of polymers. In contrast to standard spinning techniques the flow of the spinning solution is of secondary importance, the driving force in electrospinning not being mechanical but electrostatic. The physics and chemistry involved in the process are fascinating from a fundamental research point of view, and the produced fibers have considerable potential for new devices and composite materials as they can exhibit properties that are quite unique.
The first patents related to electrospinning are now more than a century old [16, 17], with the crucial patent by Anton Formhals appearing some 30 years later [18]. In recent years there has however been a strong revival of the interest in the technique, largely because it offers a number of attractive features which fit very well with today's focus on nano-, micro- and biotechnologies and advanced materials research. First, the technique allows easy fabrication of exceptionally thin continuous fibers with diameters down to some 3 nm in extreme cases, difficult if not impossible to achieve with other spinning techniques. Second, by using different variations of the basic set-up a range of different fibers with varying functionalities can be produced, including composite fibers with different materials in the core and in the sheath, and they can be collected aligned or unaligned, the latter resulting in non-woven textiles. Third, the equipment needed is of very small-scale and—compared to the equipment for standard spinning techniques—inexpensive, such that an electrospinning set-up can easily be incorporated in a standard research lab. Finally, the understanding of the electrospinning process has dramatically improved during the last 10–20 years, largely due to seminal works, e.g., from the groups of Darrel Reneker (University of Akron), Alexander Yarin (University of Illinois at Chicago), Joachim Wenndorf and Andreas Greiner (Universität Marburg), Gregory Rutledge (MIT), Seeram Ramakrishna (National University of Singapore) and Younan Xia (Georgia Institute of Technology). The modern development of electrospinning was recently well summarized by Reneker and Yarin [3]: Electrospinning has rapidly changed fiber making from a capital intensive, large scale process to a low cost, broadly applicable method that manufactures fibers on a laboratory bench, to serve diverse needs ranging from materials science and technology to life sciences and clinical medicine.
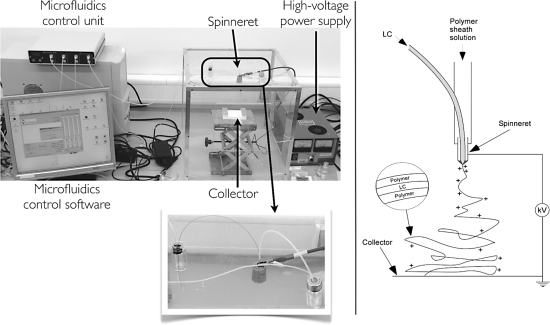
The two key components of an electrospinning set-up are a high-voltage DC power supply and a syringe pump (or two or three of them) or microfluidics control unit. One further needs containers and tubing for the fluids to be spun (when a syringe pump is used a syringe can basically fill both purposes), a metal capillary that acts as the spinneret (the nozzle through which the fluid to be spun is ejected), a target electrode or combination of electrodes on which the fibers can be collected (while this is often referred to as the collector, fiber collection is in fact often done on an object inserted between the spinneret and the target electrode), and a construction for conveniently varying the distance between spinneret and target electrode. Finally, one generally closes off a volume of air in which the spinning takes place using, e.g., a plexiglass box, in order to have some control over the atmosphere in which the fibers are spun, the relative humidity being of particular importance. Figure 7.1 shows a picture of what an electrospinning set-up can look like as well as a scheme of the fundamentals of the process.
Figure 7.1 A set-up for coaxial electrospinning using a microfluidics control unit instead of syringe pumps (left) and a schematic drawing illustrating the process.
7.2.1 Polymers and Solvents Convenient for Electrospinning
As will be explained below, the fluid to be spun needs to be non-Newtonian. The typical choice is a polymer solution although also polymer melts can be spun successfully. The solvent evaporates during the spinning process, leaving the polymer or polymer blend to form the solid fiber. Also non-polymeric fluids or particles can be incorporated within the polymer fiber by using variations of the basic technique, something we will discuss in more detail once the basic electrospinning process has been introduced.
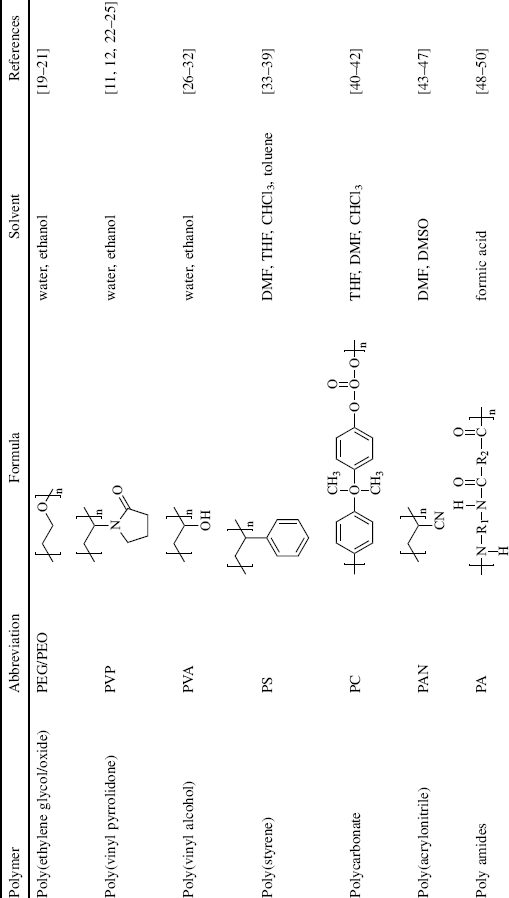
While many different polymers have been electrospun (some examples are presented in Table 7.1) using a variety of solvents, water- or ethanol-soluble polymers such as poly(ethylene glycol/oxide) (PEG/PEO), poly(vinyl pyrrolidone) (PVP), poly(vinyl alcohol) (PVA), and various cellulose derivatives are convenient to work with from a practical and environmental point of view. Fibers of poly(styrene) (PS), poly(acrylonitrile) (PAN), polylactide (PLA), and polyamides (PA) are also commonly produced by electrospinning, but they require organic solvents that may be less pleasant to work with. Molecular weights are typically in the range of 100,000 to several million and the polymer concentration is often around 5–30%, giving the spinning fluid a zero-shear viscosity similar to that of honey, about 3 Ns/m2 [3].
Table 7.1 Examples of polymers and solvents (the list is nonexclusive, solvent mixtures often being used) commonly employed in electrospinning.
7.2.2 The First Zone: Formation of the Taylor Cone
In contrast to ordinary spinning processes, the pressure on the spinning fluid is weak and the flow consequently becomes very slow in electrospinning. If no electric field is applied there will be a drop of the spinning fluid leaving the spinneret perhaps every 1–10 s. Thus, the flow is not a driving force in the spinning process; it is only required to constantly replenish the spinning solution at the spinneret. The driving force is instead an electric field applied to the fluid, typically by attaching the metal capillary to the hot pole of the DC power supply, whereas the ground is connected to the collector. The voltage applied to the capillary is very high, in the range of 10–20 kV, hence the power supply must be a rather special one (in fact, most groups doing electrospinning use the same brand of power supply: Gamma High Voltage Research). The current should however be very low (if not there is a short-circuit and something is severely wrong), hence the power supply will deliver only some hundred μA, rendering the set-up quite safe to work with. The distance between the spinneret and the collector is typically around 10 cm, so the applied field is on the order of kV/cm or 0.1 V/μm. It is thus comparable to the electric fields typically applied for switching liquid crystals, which may however also be even an order of magnitude or two higher.
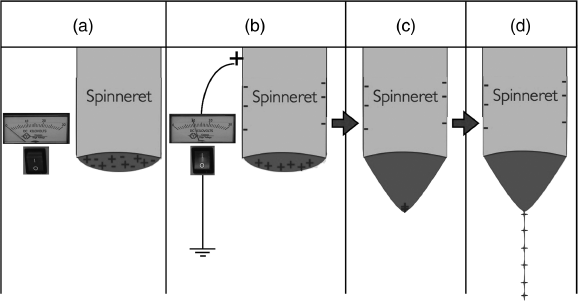
The electric field leads to a separation of charges in the spinning fluid, e.g., the positive charges accumulating at the surface of the drop protruding from a positively charged spinneret (or negative charges for a negative spinneret voltage), as illustrated in Figure 7.2b. The charges in question are typically ions dissolved in the spinning solution, in some cases deliberately added in order to ensure sufficient charge density. With the application of the electric field a conflict between surface tension and electrostatic forces arises: while the former promotes a spherical droplet shape in order to minimize the surface of the liquid, the latter promotes a movement of the positive ions closer to the collector. The result is a deformation of the droplet into a cone shape (Figure 7.2c), referred to as the Taylor cone after the British physicist and mathematician Sir Geoffrey Ingram Taylor. Once the electric field is sufficiently high the electrostatic repulsion between the ions at the cone tip, and the attraction of the ions towards the collector, are so strong that they overcome the surface tension, leading to a liquid jet shooting out from the Taylor cone [3], as illustrated in Figure 7.2d. This is the basis of electrospinning (the region with the capillary orifice and the Taylor cone can therefore be called the first zone of electrospinning) as well as of a technique referred to as electrospray. We will say a few words about electrospray and the distinction from electrospinning below.
Figure 7.2 The different steps in the formation of the Taylor cone and jet ejection, the first stage of electrospinning. (a) The spinning fluid is slowly pumped through a capillary (the spinneret) until it protrudes slightly from the capillary orifice. (b) The electric voltage applied to the spinneret separates the ions in the fluid, those with the same charge as the voltage being repelled towards the boundary of the protruding drop. (c) These charges are attracted so strongly towards the collector that the drop deforms into a cone. (d) Eventually the self repulsion between the accumulated charges and their attraction towards the collector overwins the surface tension of the fluid, leading to the ejection of a thin jet of highly charged fluid from the cone tip towards the collector.
Some delay between field application and jet formation may occur, the magnitude depending on the rheological properties of the spinning solution. In typical cases, it is less than a second but with a highly viscoelastic spinning fluid such as a polymer melt the jet ejection can be delayed by as much as 90 s [3]. The size of the orifice has little impact on the jet diameter when it leaves the Taylor cone, as long as it is not so small that it leads to viscous flow restriction [3]. The optimum pumping flow rate is the one exactly equal to the rate at which the jet carries the fluid away from the capillary during spinning. As this increases with the applied electric field, the pumping flow rate must be adjusted accordingly if the voltage is changed.
7.2.3 The Second Zone: Jet Formation or Electrospray
If the spinning solution would be water or another ordinary Newtonian liquid, the jet would soon break up into droplets as a result of the Rayleigh instability (also referred to as capillary instability) that minimizes the surface energy of the liquid. When this happens we have electrospray, a technique that has become very important at the sample preparation stage of mass spectrometry, in particular in the study of large biomolecules such as proteins (John B. Fenn was awarded the 2002 Nobel prize for chemistry for the introduction of “electrospray ionization” into the field of mass spectrometry). The division into droplets leads to rapid evaporation of liquid and a consequent strong increase in charge density within each droplet, quickly becoming so high that the initially formed droplets break up into much smaller ones, a process referred to as “atomization” (it is physically analogous to the initial formation of the Taylor cone).
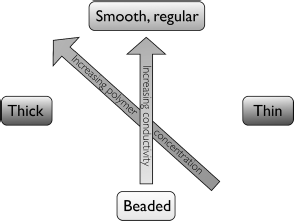
Electrospray is something we wish to avoid in electrospinning and we do this by using non-Newtonian liquids as spinning solutions. As mentioned above they are most often solutions or melts of polymers with very high molecular weight. The viscoelastic properties of the polymer solution/melt counteract the separation into drops due to the Rayleigh instability and stabilize the jet, which thus stays intact and moves towards the collector. Nevertheless, the Rayleigh instability may be partially effective, producing a chain of regularly spaced drops (beads) along the fiber. This morphology is normally not desired and one must then modify the spinning parameters to regain continuous, smooth fibers. To some extent beading can be counteracted by increasing the feed rate of the spinning solution but more effective is to modify the composition of the solution. If one cannot switch to a polymer with higher molecular weight one can either raise the polymer concentration, although this will also give thicker fibers, or increase the conductivity of the solution, e.g., by salt addition. Since thin fibers are typically desired the latter option is often to prefer. The situation is graphically summarized in Figure 7.3.
Figure 7.3 The character of the produced fibers can be tuned between “beads on a string” and smooth fibers with constant thickness by varying the composition of the spinning solution. The polymer concentration can be changed (thereby also influencing the fiber thickness) and/or the solution conductivity may be raised (e.g., by salt addition).
Directly after leaving the Taylor cone the acceleration of the fluid in the electric field is so strong that the jet acquires translation velocities of about 5 m/s [3]. This should be contrasted with the typical drift speeds of the ions in the solution on the order of 0.1 m/s [56] and the very slow flow rate of the fluid(s) involved. This means that translational flow as well as charge translation within the jet can essentially be neglected from the point where the jet leaves the Taylor cone. In a first approximation, we can consider the electrospinning process as pure elongational flow and the relevant viscosities of the involved fluids are thus the elongational rather than the shear viscosities.
7.2.4 The Third Zone: Bending Instabilities and Fiber Stretching
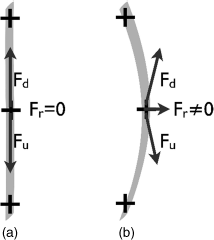
On the way from the spinneret to the collector the jet is longitudinally stretched due to the repulsion between the like-charged ions that it carries with it. It also repeatedly experiences a second instability which is of great importance for the electrospinning process and the resulting fibers [3, 57]. This is a bending instability, also fundamentally resulting from the electrostatic repulsion between like-charged elements along the jet, but now in conjunction with a lateral fluctuation. As illustrated in a highly simplified way in Figure 7.4, considering only three charges distributed along a certain length of the jet, the electrostatic self repulsion will lead to no net force on the central element as long as the jet is absolutely straight. The forces from the upper and lower charges (Fu and Fd, respectively, with u for “up” and d for “down”) are equal and oppositely directed; thus they compensate each other such that the net electrostatic force Fr = 0. But with a slight lateral fluctuation (Figure 7.4b) the forces are no longer balanced, yielding a nonzero lateral Fr. This amplifies the fluctuation and thus eventually giving rise to the bending instability, occurring when Fr is strong enough to overcome the elastic and viscous forces which promote the linear jet path (which is the shortest). The process is analogous to the Euler buckling process in mechanics (and thus also to the Frederiks transition, at the core of most electrooptic applications of liquid crystals), where a pillar compressed along its axis eventually breaks with a lateral movement as a lateral fluctuation is amplified by the axially applied force.
Figure 7.4 A simplified model for explaining the bending instability at the core of the exceptional stretching of the jet during electrospinning (see main text for explanation).
The bending instability gives rise to a spiral-shaped continued path of the jet with a conical envelope, cf. Figure 7.5, first with a long pitch and relatively large (and increasing) spiral diameter. Eventually a second bending instability will typically appear, resulting in a smaller diameter spiral modulation of the jet, and this can go on to a third and fourth or even fifth hierarchical instability [3, 57]. The reason for the multiple bending instabilities is the continuously increasing charge density during the flight of the jet; although one might at first think that the substantial stretching would reduce the local charge density in the jet one must remember that the rapid evaporation of the solvent reduces the total jet volume, generally leading to a net increase in charge density. As a result of the multiple bending instabilities the jet follows a very complex path from the spinneret to the collector, with a path length that is orders of magnitude greater than the actual spinneret–collector distance, and the continuous stretching along this path renders the jet very much thinner at the end of the process than at the beginning. This is what allows such exceptionally thin fibers to be produced by electrospinning.
Figure 7.5 A simple cartoon of the envelope of the jet during electrospinning, characterized by multiple superposed spiral modulations due to subsequent electrostatically induced bending instabilities. Redrawn from [3].

The elongation of the jet due to the multiple bending instabilities will normally continue until it hits the collector (or an object inserted in the spinning path) or until the polymer solidifies due to solvent evaporation or, in case of melt spinning, to cooling. During the elongation stage the jet experiences stretching ratios in the range of 105 and stretching rates up to 105/s, values which are unmatched by other methods like fiber extrusion followed by mechanical stretching [6].
7.2.5 The Fourth and Final Zone: Fiber Collection
Just before the jet reaches the collector its translational speed is very high, often on the order of 40 m/s [6]. As it hits the collector the motion is abruptly stopped although the impact is not necessarily quite what one would expect considering the speed. Because of the bending instabilities during the jet flight the motion is largely lateral, in the plane of the collector, and the fiber can therefore be deposited as a relatively straight fiber. Depending on the vertical position of the collector, the impact of the jet may however be more or less head on, and in that case the buckling instability arising upon impact will lead to a characteristic small-diameter spiral pattern of the deposited fibers [3].
Normally one tries to optimize a set-up such that the jet solidifies just before or just after it hits the collector. If it is too wet the fiber morphology is not stable and deposited fibers will merge as a result of flow after deposition. If it is entirely dry then the decharging of the fiber via the contact with the grounded collector takes longer time. Since the fiber is still highly charged when it lands on the collector it repels the jet that approaches from above, which therefore moves laterally towards a different collection spot. As long as the charge on the deposited fiber is quickly neutralized this is fine; it results in a criss-cross deposition of fibers producing a characteristic open network mat structure, cf. Figure 7.6a. But if the decharging takes too long the repulsion of the incoming jet may be so strong so that the fibers are instead distributed in an uncontrolled way around the target area, building bridges between various surfaces around the collector.
Figure 7.6 Scanning electron microscopy images of coaxially electrospun fibers with a nematic liquid crystal core and a composite sheath of PVP and TiO2. The fibers were collected as a random non-woven mat (a) and aligned between the arms of a tweezer (b). Images courtesy of J. McCann and J. Lagerwall.

In many cases an aligned collection of fibers is desired and a number of approaches have been developed to achieve this. The most obvious, but practically more involving, solution is to collect the fibers on a quickly rotating drum [58, 59], leading to fibers aligned perpendicular to the rotation axis. Quite well aligned fibers can however be achieved in a much easier way, simply by splitting up the collector electrode in two parallel grounded plates or wires [22, 50]. The electric field profile now has a horizontal component that promotes stretching of the fibers across the gap between the electrodes. Together with the repulsion between fibers that are suspended between the electrodes, thus only slowly discharged, this leads to an alignment of the fibers across the gap that can be quite good. The simplest way of realizing this type of alignment is to put a pair of metal tweezers on top of a flat collector electrode, resulting in the fiber jumping back and forth between the arms of the tweezers. The aligned fibers in Figure 7.6b were obtained in this way.
An interesting variation of this alignment technique was introduced recently by Yan et al. [60]. Rather than splitting up the target electrode in two, they placed two parallel blocks of a high permittivity dielectric material on top of the single grounded target electrode, again resulting in a lateral component of the electric field that stretches the fibers across the gap between the dielectric blocks. They demonstrated the concept with PVA fibers spun from aqueous solution onto water containers, blocks of wood, or ferrite, the latter yielding the best results.
If control of fiber alignment is much more critical than reduction of fiber diameter an attractive alternative can be to terminate the flight of the jet prior to the first bending instability, i.e., the third zone is eliminated from the process. Using this so-called near-field or high precision deposition electrospinning, in which the spinneret–collector distance is often only about a millimeter [61–63], excellent control of the position and alignment of the fibers can be achieved. Moreover, by using a collector substrate movable by an xy-translator very sophisticated patterns can be spun. (In principle also the spinneret could be moved but this is practically much more challenging since the spinneret is connected to the tubes for feeding with spinning solution as well as to the hot pole of the power supply.) This comes at the cost of somewhat thicker fibers and probably poorer alignment of the polymer chains within the fiber, since the extreme stretching induced by the multiple bending instabilities has been removed from the process. Moreover, the fibers may still be deposited with local spiral morphology due to a buckling instability at impact on the collector, especially for spinning distances greater than 1 mm [61].
7.2.6 Coaxial Electrospinning
A version of electrospinning that is particularly useful for incorporating low molar mass liquid crystals (or other liquids not amenable to spinning) is coaxial electrospinning, where one fluid is spun inside another one [64, 65]. As illustrated in Figure 7.1 a thin capillary is introduced inside the main tubing through which the polymer solution flows. This inner capillary runs almost coaxially with the main capillary until the orifice, where the two capillaries should end at the same height. They must be well centered at this point in order for the Taylor cone to form symmetrically, with the fluid pumped through the inner capillary uniformly surrounded by a layer of polymer solution. Since the introduction of the inner capillary through the wall of the main tubing often prevents perfect coaxial alignment of the capillaries, one may need to slightly bend the metal capillary close to the orifice, such that they are well centered at the exit point of the spinneret. Alternatively, a special coaxial spinneret with dedicated inlets for the two fluids can be fabricated in a workshop.
The beauty of coaxial electrospinning is that any fluid with suitable viscosity that is immiscible with the polymer solution (jets produced with miscible fluids do not maintain the coaxial geometry throughout the electrospinning process [64]) can be spun into fibers by pumping this fluid through the inner capillary. The polymer forms a protective and confining cylindrical wall around the inner fluid. In other words, coaxial electrospinning produces fibers with a core–sheath structure, and because the sheath is a solid polymer the core can be a low molar mass liquid (it can of course also be a polymer), in our case a thermotropic liquid crystal. The inner fluid can give the composite fiber new functionality, e.g., special optical properties from a liquid crystal core (to be discussed in detail below). But even simple alkanes inside the fibers can functionalize the produced non-wovens into phase change materials useful for temperature stabilization [25].
Although we could in principle charge both or either liquid in coaxial electrospinning by using a metal capillary at the end of the inner tubing, upon connecting this to the hot pole of the power supply rather than the outer capillary (which in this case could be made of glass, for instance), the most common configuration is one where only the outer fluid is in direct contact with the electrode (the outer capillary, made of metal) and thus only this fluid gets charged. The inner fluid then does not experience the electrostatic forces that drive the electrospinning process, these being active only in the outer fluid. The jet core is instead stretched only by mechanical means as a secondary effect from the electrostatic stretching of the sheath [6]. This is advantageous when spinning living matter, such as cells, which may not survive the strong electrical charging, but it may also be a means of spinning highly conductive solutions, e.g., carbon nanotube suspensions or conductive polymers. These would short-circuit the set-up and thus terminate spinning as soon as the jet makes contact with the collector if spun as the outer (or only) fluid.
7.3 Electrospinning of Liquid Crystal Polymers
The most straightforward approach to introduce liquid crystallinity to electrospinning is to work with liquid crystalline polymers for creating the fibers. These can be of thermotropic liquid crystal type, i.e., they form a liquid crystalline state after the solvent has evaporated or the isotropic melt has cooled down during the spinning process, resulting in a final fiber product that is liquid crystalline (possibly at elevated temperature). Also lyotropic liquid crystal polymers can be spun, in this case forming, e.g., a nematic phase during the spinning process, while the solvent is present. The final solvent-free fibers are then crystalline. Both approaches have been explored.
Canejo et al. investigated electrospinning from a lyotropic liquid crystalline solution of acetoxypropylcellulose (APC) in dimethylacetamide (DMAc) [14]. The concentration was 60 wt% concentration, rendering the mixture cholesteric, yet this was near enough to the isotropic regime to ensure a viscosity that was not too high for spinning. The fiber product had some slightly unusual characteristics both on the large and the single-fiber scales. Rather than the typical criss-cross mat of reasonably long fibers that is usually obtained in electrospinning without alignment control, the fibers formed a web with an almost foam-like character of “cells” of varying sizes. Zooming in to the individual fibers, which were birefringent (revealing alignment of the polymer chains) and of a fairly large average diameter of just below 3 μm, the authors found these to be helically twisted. Interestingly, both right-and left-handed twists occurred (in a 3:1 ratio), hence the twisting is not a direct expression of the chirality of the polymer. Nevertheless, the theoretical analysis in the paper concludes that the chiral liquid crystalline state and its response to the electric field that drives the spinning process are at the core of the fiber twisting.
Solutions of a biphenyl-based smectic-forming thermotropic liquid crystalline main-chain polymer (molecular weight 29,000g/mol) in hexafluoropropanol were electrospun by Nakashima et al. [66]. By adding the organic electrolyte pyridine at varying concentrations the team could modify the conductivity of the spinning solution, thereby tuning the diameter of the resulting fibers from about 100 nm to 5 μm. The internal polymer chain organization was characterized by X-ray scattering, revealing that fibers with a diameter greater than or equal to 220 nm exhibited a smectic structure. This was of a polydomain character in the thickest fibers but for a fiber diameter below 3 μm the layers were aligned perpendicular to the fiber axis. The thinnest fibers, with a diameter slightly above 100 nm, did not reveal any sign of chain ordering in the X-ray diffractograms.
Surprisingly, two different layer spacings were found for the smectic fibers, 24.0 Å and 17.0 Å. The authors explained this observation by the fact that the thermodynamically stable state of the polymer at the temperature of characterization is the SmCa phase, characterized by tilted mesogen units, in opposite directions in adjacent layers. The tilt reduces the layer spacing compared to a non-tilted phase, like SmA, resulting in the smaller layer spacing of 17.0 Å. However, the strong stretching of the polymer chains in the electrospinning process promotes a metastable SmA state, yielding the larger 24.0 Å layer spacing. Annealing the fibers at 50°C (greater than the glass temperature Tg = 30°C) gradually transforms the SmA phase regimes to SmCa and thus only the 17.0 Å layer spacing diffraction is observed after annealing.
The liquid crystalline polymer Vectra was melt-electrospun, thus without the use of solvent, by Ogata et al. [67]. Their modified set-up had a rod of Vectra being slowly translated towards the spinneret, where the rod tip was strongly heated by a triplet of CO2 lasers, such that the viscosity of the polymer reached low enough values for electrospinning. The distance between spinneret and collector was rather small, only 2.5 cm. The small distance and the absence of solvent lead to relatively thick fibers, about 2.5 μm, but the chains were quite well aligned along the fibers, judging from the X-ray scattering diffractogram.
Considering the strong stretching during electrospinning, normally ensuring a highly uniform alignment of the director along the fiber, a very appealing concept is to produce liquid crystalline elastomer fibers by electrospinning, the crosslinking taking place during or immediately after spinning for fixating the uniaxial order. By heating the fiber to the clearing point of the liquid crystalline elastomer, strong mechanical actuation effects could be expected. As the polymer chains acquire a more or less random coil conformation in the isotropic phase the fiber length should be reduced and the diameter increased [68]. In a joint effort by the Wendorff/Greiner and Finkelmann teams, Krause et al. explored this concept using a main chain copolymer comprising a mesogenic and a photocrosslinker monomer, respectively [15]. The polymer, with a molecular weight of Mn ≈ 44,000 g/mol, was dissolved in chloroform at a concentration of about 7% by weight and spun with the relatively large spinneret–collector distance of 19 cm, at a consequent high voltage of about 25 kV.
The produced fibers, with an average diameter of about 1.5 μm, were birefringent, confirming the uniaxial alignment of the director along the fiber axis. Surprisingly, the report contains no discussion of the effect of heating the fibers above the clearing point. Obviously further studies along this interesting line of research are needed, but the lack of discussion of any actuation from the fibers gives a hint that the concept is not as straightforward as one may initially expect. Possibly the fact that the molecular weight is still rather low compared to typical polymer samples used for electrospinning renders the fiber production difficult.
The insufficient molar mass of the liquid crystalline polymer seems to have been the main reason for the difficulties encountered also in the study by Wu et al., investigating electrospinning of a polysiloxane backbone polymer with cholesteryl mesogen side chains [69]. Although the polymer concentration was very high (33% in chloroform), electrospinning did not succeed using this polymer on its own. Only upon addition of 5% of high molar mass poly ethylene oxide (PEO, Mn = 1,000,000), mixed into the chloroform solution of the liquid crystalline polymer, did high quality fibers result. The fiber diameter could be kept quite low, between 0.5 and 2 μm. Alternatively, some improvement of the fiber quality was obtained upon addition of low molar mass liquid crystal, an observation that is not so easy to explain. The improvement was however not sufficient and the fiber diameter also increased strongly in this way, rendering the mixing with PEO the better choice.
Interestingly, data from X-ray, scanning electron microscopy (SEM), and differential scanning calorimetry (DSC) investigations revealed no sign of phase separation between the liquid crystal polymer and the PEO, but they seem to have formed a homogeneous blend in the final fibers. Directly after electrospinning the polymer chains were aligned along the fibers, but not as well as might be expected, even worse than after conventional drawing. The authors attributed this observation to the fast solvent evaporation during spinning. By annealing the fibers for 20 min at 70°C the degree of chain orientational order went up considerably.
In summary, the experiments so far on electrospinning of liquid crystalline polymers show promise and interesting observations have been reported. Some problems still remain, however, most likely related primarily to the difficulties in realizing liquid crystalline polymers with very high molecular weight. By mixing the liquid crystalline polymer with regular, classically electrospinnable polymers, quite good results can nevertheless be achieved.
7.4 Low Molar Mass Liquid Crystals Inside Electrospun Fibers
Not only somewhat more complex but also much more versatile due to the immense catalog of available materials, is the spinning of low molar mass liquid crystals. The trick is to spin the non-polymeric, and thus “un-spinnable”, liquid crystal as a core fluid contained inside a polymer sheath solution. The two approaches that have been explored for achieving this situation is to either mix polymer and liquid crystal in a common solvent and then rely on phase separation during the spinning process for obtaining the core–sheath structure [9], or to use coaxial electrospinning to establish the core–sheath geometry already at the starting point [10–12]. The resulting core–sheath fibers constitute an entirely new configuration for studying—and applying—low molar mass liquid crystals, opening possibilities very different from standard device configurations. The liquid crystal gives the fibers new functionality and responsiveness and the encapsulation of the liquid crystal in the narrow cylindrical fiber core allows easy investigation of 1D confinement effects. The fibers also have large application potential. While we are still at the very beginning of the research on development of liquid crystal-functionalized electrospun fibers and non-wovens one can envisage applying them in fields ranging from full spatial resolution temperature sensing, via dynamic camouflage clothing, to wearable electronics and displays. For simplicity we will in the following refer to low molar mass liquid crystals simply as “liquid crystals”.
7.4.1 Non-Coaxial Electrospinning of Liquid Crystal Core Composite Fibers
Buyuktanir et al. devised an approach to liquid crystal electrospinning that is very attractive through its simplicity and the fact that a standard electrospinning set-up can be used. The commonly studied nematic liquid crystal 5CB was dissolved together with polylactide (PLA, Mw = 186,000g/mol) in a chloroform–acetone solvent mixture (3:1 volume ratio), in proportions giving a PLA concentration of 7.3 wt%. This polymer concentration was kept fixed while the proportion of 5CB to PLA was varied in the range of 7–86%, spinnable solutions being those with 5CB concentrations below 70%. During the spinning process the 5CB phase separates from the polymer, forming the liquid crystal core of the fibers.
The phase separation between 5CB and PLA is however not complete but the fiber sheath actually becomes a mixture of PLA and 5CB. The authors found that phase separation started at 5CB to PLA ratios greater than 28 wt%, meaning that the excess 5CB then started forming a polymer-free liquid crystal core within the mixed composition sheath. Above this liquid crystal fraction the clearing transition enthalpy peak could be detected in differential scanning calorimetry measurements and the fibers appeared clearly birefringent for more than 40% 5CB, with the director generally aligned along the fibers as detected by polarizing microscopy. The morphology of the fibers (with diameters in the range of 0.5–2 μm) was however rather beaded for the liquid crystal-filled fibers.
7.4.2 Coaxial Electrospinning of Fibers with Solvent-Free Liquid Crystal Core Fluid: Practical Requirements
While the above approach is practically straightforward to realize, as only a single solvent-rich isotropic liquid phase is being spun, the degree of control is quite limited. The fiber beading in the report described above was also quite significant. It may thus be a worthwhile investment to modify a set-up for coaxial electrospinning to allow spinning of the pure liquid crystal as the core fluid, without addition of solvent, ensheathed by the outer polymer–solvent solution. While it should be possible to spin both thermotropic and lyotropic liquid crystals in this way the experiments carried out so far have been restricted to thermotropics. With lyotropics the situation obviously gets more complex as solvent is then a critical component of the liquid crystal phase, hence both sheath and core fluids contain solvent which may or may not evaporate during or after the spinning.
Generally, the coaxial approach to liquid crystal electrospinning requires the basic set-up to be extended with a tailored temperature regulation system, allowing the user to choose the phase adopted by the liquid crystalline material at the onset of spinning. While nematics can be relatively easy to electrospin, the shear viscosity η2 in fact being even lower than the shear viscosity of the material heated to its isotropic phase, higher-ordered liquid crystal phases such as smectics or cholesterics are often (but not always) too viscous. It may be difficult to achieve continuous and smooth flow to the spinneret of a highly viscous fluid. Moreover, a large difference in elongational viscosity between inner and outer fluids complicates the spinning process such that a continuous filling of the fibers may be impossible. It is thus highly desirable to have the possibility to heat the material into the isotropic phase if we wish to prepare fibers with a core that eventually is in a high-ordered liquid crystalline state.
It is not trivial to realize a precise and versatile temperature control for electrospinning, since the substance to be heated is spatially distributed over the full length of tubing, the access for a heating system being very difficult in certain places. The simplest solution is to heat the whole system up to the spinneret with a heat gun. This method is readily accessible but obviously lacks precision, preventing true control of the spinning parameters and making repeatability challenging, and it also heats many components that would better stay at room temperature, such as the syringe pump in case this device is used. If instead a pneumatic microfluidics system is used for pumping the fluids this has the advantage that the vial from which the liquid crystal is pumped can easily be placed in a heating block for temperature control. This improves the situation but it is often not sufficient, since the large surface area of the tubing through which the liquid crystal flows renders temperature equilibration with the surrounding air very rapid. Despite heating the fluid while in the reservoir it may thus end up too cold once it reaches the spinneret. A design that proved quite effective is one where a heating block for the liquid crystal vials is combined with a heated air flow along the tubing. Even more efficient temperature control of the tubing may be realized by immersing it in a heated liquid bath, although this obviously places quite stringent requirements on sealing the liquid bath where the tubing leaves the bath, just before the spinneret and the electrical connection to the power supply.
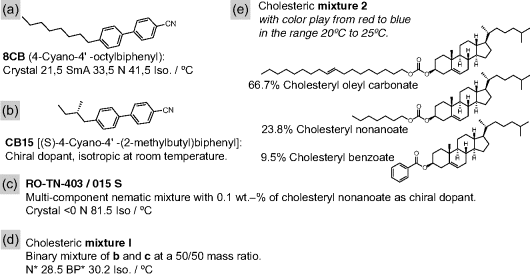
For some liquid crystal substances the inner fluid can in fact sometimes be in a cholesteric or even smectic phase during spinning, rendering the heating requirements less critical. For instance, 8CB (Figure 7.7a) could be spun at room temperature where the liquid crystal is in a SmA phase, close enough to the second-order N-SmA transition that the material still flows reasonably easily. Likewise, the short-pitch cholesteric formed by doping the commercial nematic mixture RO-TN-403/015 S with the chiral dopant CB15 (see Figure 7.7b–d) had sufficiently low viscosity to be spun at room temperature. This mixture (in the following referred to as mixture 1) is much less viscous than the three-component cholesteryl ester mixture 2 (Figure 7.7e) although both have about the same very short pitch of the director helix at room temperature.
Figure 7.7 Key substances used for preparing the liquid crystal core materials in the electrospun composite fibers studied by the author's team.
With other liquid crystals strong heating is required for electrospinning, or one may have to resort to dissolving the liquid crystal in a solvent also in this case, being careful to use a solvent that is not miscible with the sheath solution and that will evaporate to a large extent during the spinning process. This is for instance the case when the clearing point is higher than the maximum spinning temperature of the sheath solution (often quite low due to the requirement to use reasonably volatile solvents in this solution) or when even the isotropic phase is too viscous, as can be the case for some discotic, bow-shaped, or oligomeric liquid crystals.
7.4.3 Sheath Morphology of Coaxially Spun Liquid Crystal Core Fibers
The first few experiments on coaxial liquid crystal core–polymer sheath electrospinning were carried out with an outer spinning solution containing not only just polymer but also a Ti(OiPr)4 sol–gel precursor [12]. The result is a polymer–TiO2 composite sheath that is particularly stable, making coaxial electrospinning with core fluids that are close to Newtonian relatively easy. The exact composition of the sheath solution was in this case 3% PVP, 29% Ti(OiPr)4, 48% ethanol, and 19% acetic acid, yielding a sheath where the polymer is in fact just a minority (27%, the rest TiO2). While this gave very stable fibers with well-defined morphology (Figure 7.6) the fact that the sheath consists mainly of TiO2 makes characterization of the liquid crystal core more challenging, and the optical properties of the liquid crystal are inaccessible behind this strongly scattering and birefringent sheath. The verification of the director alignment as well as the investigation of the phase sequence therefore had to be done with polarized Raman spectroscopy.
In later work the fibers were spun with a pure PVP sheath that is largely non-birefringent. This generally worked very well, fibers produced using a 12.5% PVP in ethanol solution as outer fluid appearing quite uniform in optical microscopy and the liquid crystal optical properties being easily detectable (these will be discussed below). The outer morphology of these fibers was investigated by SEM; some results were shown in Figure 7.8. The fibers were generally cylindrical although some examples of essentially flat ribbons were also found. Depending on the degree of liquid crystal filling, the outer diameter ranged between one and a few microns.
Figure 7.8 Scanning electron microscopy images (courtesy of G. Scalia and O. Calò) of pure PVP sheath fibers with cholesteric liquid crystal core (mixture 1, Figure 7.7d). Both samples were spun at room temperature with 25–35% relative humidity, 10 cm spinneret–collector distance, and 10 kV spinning voltage, with a 12.5% PVP in ethanol solution as the outer fluid. This was pumped at a flow rate of 1.8 mL/h while the inner fluid was pumped at flow rates of 0.14 mL/h (a) and 0.36 mL/h (b).

The fact that coaxial spinning with liquid crystal core and pure PVP sheath can produce stable fibers is actually somewhat surprising, because earlier attempts by Li and Xia to spin mineral oil coaxially inside a PVP ethanol solution failed due to structural breakdown of the coaxial jet. The result was complete separation into oil droplets and solid fibers of pure PVP [64]. The addition of the Ti(OiPr)4 sol–gel precursor in the outer fluid was in their case required to stabilize the sheath. In the case with a liquid crystal in the core the situation is however somewhat different because the inner solution is also weakly non-Newtonian. Relatively long filaments can be drawn from liquid crystals, in contrast to isotropic Newtonian fluids where the Rayleigh instability more rapidly destabilizes a filament and separates the fluid into droplets. It thus seems that this property of the liquid crystal is beneficial for coaxial electrospinning, allowing us to use a simpler and in many respects practically preferable sheath solution than had the inner fluid been isotropic. This means that one should not heat the liquid crystal into its isotropic phase for spinning if it can be avoided. The nematic phase ought to be the optimum phase for spinning but unfortunately not all materials exhibit this phase.
The results of Li and Xia suggest that when encapsulating non-nematic materials a mechanically stronger sheath solution may be required to stabilize the coaxial jet. Different variations of the sheath solutions in this direction were therefore explored also for liquid crystal electrospinning, keeping the requirement that the sheath should be at least largely non-birefringent and transparent. One approach is to increase the polymer concentration, yielding fibers that are mainly cylindrical and intact, in some cases also yielding porous fiber sheaths. An alternative modification is to nevertheless add Ti(OiPr)4 sol–gel precursor but at a rather small amount, just sufficient to stabilize the sheath but not so much that birefringence and scattering from TiO2 grains become disturbing. The fibers formed with this sheath solution were truly excellent in terms of uniformity and cylindrical shape, cf. Figure 7.9, and the small amount of TiO2 in the sheath did not prevent the liquid crystal from being visible through the fibers. A further interesting observation was that the TiO2–PVP composite fibers were substantially thinner than those without TiO2.
Figure 7.9 Scanning electron microscopy images (courtesy of G. Scalia and O. Calò) of fibers with composite sheath, consisting of 91% PVP and 9% TiO2, and cholesteric core (mixture 1, Figure 7.7d). The ethanol-based sheath solution containing Ti(OiPr)4 as sol–gel precursor in addition to 10% PVP was pumped at a flow rate of about 2 mL/h and the liquid crystal core fluid at 0.26 mL/h (a) and 0.80 mL/h (b), respectively. Both samples were spun at room temperature, with 25–35% relative humidity, 10 cm spinneret–collector distance and 10 kV spinning voltage.

It is obviously of great interest to know also the internal structure of the fiber, the inner diameter being the most important parameter to establish. This is however not trivial to realize experimentally since the fibers are generally too thick for transmission electron microscopy (TEM) imaging. In addition, the similar electron density of the sheath polymer and the liquid crystal would lead to too weak contrast for distinguishing the two regimes in TEM. One approach that has been successful in the characterization of similar systems is to carry out SEM on the fractured end surface of fibers that have been broken in two sections, at best after having dissolved the core fluid with a suitable solvent that does not affect the sheath. This however requires that the fiber sheath does not collapse during the fracture, something which turned out to be very difficult to avoid for pure PVP sheath fibers.
The approach that eventually brought success in imaging the internal structure was to use Focused Ion Beam (FIB) milling to cut sections through the fibers, either along or across them, and then image the dissected fiber using SEM or FIB. When exposed to the vacuum of the FIB chamber the low molar mass liquid crystal molecules of the core evaporate, leaving only the polymer sheath to form the image. As can be seen in Figure 7.10 this results in a very clear picture of the fiber morphology, internal as well as external. The channel that initially held the liquid crystal is easily seen. In this particular fiber, rather thick and with high degree of liquid crystal filling, the morphology is not cylindrical but rather one of a horizontal cylinder segment. Apparently the soft fiber sheath had collapsed when the fiber hit the substrate. The liquid crystal core channel has roughly the same cross-section as the fiber as a whole. The FIB characterization study is described in full detail in a paper soon to be published (authors E. Enz, V. La Ferrara, O. Calò, J. Lagerwall and G. Scalia).
Figure 7.10 A richly filled fiber with pure PVP sheath and cholesteric liquid crystal core, dissected using FIB milling and imaged by the FIB. Upon hitting the substrate the fiber had collapsed from the normal cylindrical geometry into a horizontal cylinder segment morphology. The channel holding the liquid crystal can easily be seen in the regions where the FIB has cut away the sheath. Image courtesy of G. Scalia and V. La Ferrara.

7.4.4 How Does the Liquid Crystal Align inside the Fiber?
The cylindrical symmetry of the fiber (for simplicity we consider here only fibers with cylinder morphology) might intuitively suggest a director alignment along the fiber for a nematic phase but perpendicular for a SmA phase. In case of a nematic the minimum viscosity is along the director, hence an alignment along the fiber is to be expected considering the spinning conditions. Moreover, this alignment produces a defect-free director field within the cylindrical fiber, whereas homeotropic as well as planar but transverse alignment would yield very strong splay and bend, respectively, for the nematic confined in the narrow fiber core. The homeotropic alignment would on the other hand allow smectic layers to form concentrically along the fiber, compatible with easy shear flow on the way to the spinneret, promoting the hypothesis of this alignment for spinning in the smectic phase.
In case of clear and isotropic polymer sheaths the alignment of the director within the fibers is easily investigated with polarizing microscopy. Some fiber sheaths are however too scattering, or birefringent in themselves, such that this straightforward technique cannot be used for the purpose. For instance, the fibers with composite sheaths containing a large amount of TiO2 are both strongly scattering and birefringent, rendering optical microscopy investigations useless for assessing the state of the liquid crystal core [12]. In this case, polarized Raman spectroscopy turned out to be highly useful since the liquid crystal has a very characteristic response spectrum that can easily be distinguished from that of the sheath. The strength of the liquid crystal peaks depends on the alignment of the director with respect to the polarization of the excitation laser, going to zero for perpendicular alignment and being maximal for parallel. This allows the director orientation and the effective degree of orientational order to be assessed also in such fibers.
In case the fibers have been spun with uniform fiber orientation they can also be placed in an X-ray scattering set-up, allowing the type of liquid crystal alignment in the fiber to be elucidated from the X-ray scattering diffractograms. This is particularly useful for smectic liquid crystal cores, since we can then study the director alignment and the layer formation independently. A convenient approach is to roll up an aligned and relatively dense fiber mat into a scroll, perpendicular to the fiber axis, which can then be inserted into an X-ray capillary for the experiment. An example of the result for a SmA-containing fiber is shown in Figure 7.11, the scroll production illustrated on the left and the resulting 2D X-ray pattern shown to the right.
Figure 7.11 X-ray diffraction pattern (right) from 8CB inside coaxially electrospun PVP fibers, rolled into a scroll (illustration on the left) and placed into a Mark capillary. From paper [11].

It turns out that the assumption about the nematic orientation is correct (even for chiral nematics), but for smectics the layers do not form concentrically but perpendicular to the fibers. Even in the smectic phase planar alignment prevails, resulting in a stack of smectic layers along the fiber, each layer with a lateral extension no greater than the fiber diameter. The diffractogram in Figure 7.11 features clear small-angle smectic layer diffraction peaks along an axis perpendicular to the rolling direction, thus along the fiber axis. A more careful consideration of the situation makes this observation understandable. Since the flow during the actual spinning stage is of elongational type it is the elongational and not the shear viscosity that is relevant. For smectics the lowest shear viscosity is perpendicular to the director and along the layers, since the layers can slide along each other. But the elongational viscosity in this direction can be expected to be rather high, since all layers are stretched together, without flow past each other, in elongational flow. Elongational flow along the director may be preferable since the elongation can easily be accommodated by making the lateral extension of each layer somewhat smaller and moving the molecules from the sides of the cylinder to the middle in order to form new interstitial layers. Finally, this smectic geometry may also be promoted by the fiber sheath imposing a strong planar director alignment at the boundary between liquid crystal and fiber sheath.
The lateral extension of the smectic layers is very small with this geometry, only the ≤1 μm diameter of the confined space inside the fiber. This size is smaller than the thickness of the thin cells typically used for preparing planar surface-stabilized ferroelectric liquid crystal (SSFLC) samples of very short-pitch chiral SmC* materials. In that case, a geometry with small lateral smectic layer extension, albeit only in one dimension, is secured by means of strong director anchoring at the sample boundaries. For the cylindrical symmetry of a core–sheath fiber the situation is however different, in a quite interesting way. While the geometry with the director along the fiber axis is unproblematic for a SmA phase, the tilted director of SmC* would lead to frustration. For a non-chiral SmC phase the requirement that each smectic layer should have uniform director tilting direction forces the layers to tilt in a uniform direction. Going to a chiral SmC* phase, however, this solution fails since the director tilt precesses along the layer normal in this phase, hence each layer would need to tilt in a direction slightly different from its neighbor layers above and below. Obviously this is incompatible with the stacking of the layers.
It is thus a very interesting question for future experiments to establish how an SmC* phase resolves this frustration. One may consider a number of scenarios. First, the SmC* phase may be replaced by SmA* in order to avoid the problem. Second, the helix may be expelled as in flat SSFLC samples, but here all layers would be uniformly tilted as in the geometry expected for a non-chiral SmC phase within the fiber. This would yield the fiber spontaneously polarized with a component perpendicular to the fiber, probably with an impact on the actual spinning process in the electric field and obviously something that could be exploited in applications. Third, a modulation of the director tilt magnitude may occur on approaching the core perimeter, the tilt disappearing into a SmA-like geometry at the border of the fiber sheath. The larger part of the interior of each layer would then still be in SmC* geometry with a well-defined tilt direction within each layer (which would be perpendicular to the fiber axis) that precesses along the layer normal. Of course other scenarios are possible. In case of SmC* phases with high spontaneous polarization, the polarization may also play a role for the resulting alignment since the director field under confinement has been shown to be strongly influenced by high polarization values in case of planar cells [70].
7.4.5 Phase Sequence and Phase Transitions of Liquid Crystals Inside Electrospun Fibers
It is well known that the phase sequence of liquid crystals can be strongly affected by confinement [71–75], particularly sensitive phases such as the chiral smectic-C intermediate phases even disappearing completely already at relatively large surface spacings, on the order of 10 μm [72]. As the inner sample spacing in this latter case is an order of magnitude larger than the outer fiber diameter of typical electrospun fibers, we can clearly expect to see an influence from the encapsulation, even on more robust phases like nematic and SmA. Moreover, the cylindrical symmetry may have a particular effect, as already speculated above for SmC and SmC* phases and as will be further discussed for the case of chiral nematics and blue phases below.
In the first few experiments on coaxial electrospinning with a nematic liquid crystal mixture inside a composite PVP–TiO2 sheath a dramatic increase of the clearing temperature by about 20°C compared to the bulk liquid crystal was found [12]. Moreover, the first-order character of the nematic–isotropic transition also seemed to have disappeared as a result of the strong confinement. In a series of elegant Atomic Force Microscopy investigations on liquid crystals in the vicinity of solid substrates, the team around Igor Musevic in Ljubljana demonstrated how liquid crystalline order prevails very near the surface even at temperatures well above the clearing temperature [76, 77]. In the electrospun fibers, where the liquid crystalline core is fully surrounded by a solid sheath with an inner diameter typically a few hundreds of nanometers the same phenomenon should occur. However, the molecules in the vicinity of the surface now constitute a much greater fraction of the whole liquid crystal volume than in usual samples. We may thus explain the observation of increased clearing point and the loss of first-order transition character as a result of a surface-induced stabilization of the liquid crystal phase that gets less effective on going from the sheath to the core. As we continue heating the fiber from the bulk clearing temperature the mixture gradually turns isotropic from the center, the ratio of nematic to isotropic phase continuously getting smaller until finally, some 20°C above the bulk clearing temperature, even the surface layer has lost its orientational order. In this way the phase transition appears continuous to the outside world and liquid crystalline order is detectable at much higher temperatures than for bulk samples.
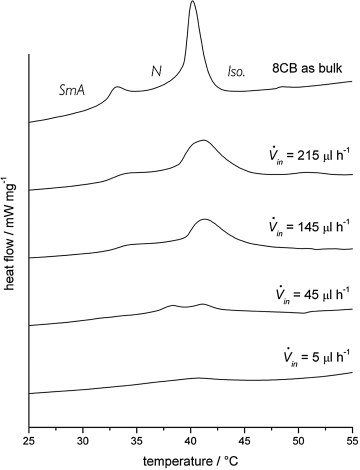
With relatively thick fiber mats prepared by coaxial electrospinning of 8CB inside a pure PVP sheath, a sufficient amount of liquid crystal was contained to give a response in DSC experiments [11]. This allowed a study of the effect of encapsulation on the SmA-N and N-isotropic phase transitions. As expected, the phase transition peaks became weaker and more smeared out than for a bulk sample, and the two transitions approached each other upon increasing confinement, until they could no longer be resolved in the thinnest fibers, cf. Figure 7.12. The increase in clearing point was here less pronounced than in the previous study, most likely because the encapsulation was not as strong in these pure PVP sheaths as in case of the TiO2–PVP composite fibers. The strong effect on the smectic–nematic transition temperature was explained as a result of an encapsulation-induced kinetic stabilization (superheating and -cooling) of whatever molecular arrangement that is in place. When first passing a certain phase transition temperature the mesogens at the inner surface of the polymer sheath are strongly anchored in an arrangement corresponding to the starting liquid crystal phase. This surface-stabilized cylindrical layer of molecules will counteract reorganization into a less or more ordered phase, leading to the strong supercooling/-heating effects.
Figure 7.12 Differential scanning calorimetry curves on heating for 8CB in electrospun PVP fibers, as a function of liquid crystal flow rate (lower rate yielding thinner fibers, with stronger confinement). From paper [11].
A somewhat different type of modification of the phase sequence was observed when the chiral mixture 1 was encapsulated in fibers. Here the phase transition between N* and BP* truly disappeared, on heating as well as on cooling. While also this effect was a consequence of the influence from the sheath on the liquid crystal structure formation, it was not a simple matter of kinetic stabilization. In fact, the two phases had become degenerate in the cylindrical confinement, as will be explained in the following.
7.4.6 The Optical Properties and Phase Behavior of Cholesteric and Blue Phases Inside Electrospun Fibers
Enz and Lagerwall prepared a series of PVP sheath fibers containing the short-pitch cholesteric liquid crystal mixture 1, reflecting a blue-green color when observed straight on [10]. In the experiments the liquid crystal flow rate as well as that of the sheath solution was varied. With increasing liquid crystal flow rate the fiber core, and thus the fiber, gets thicker and the sheath thinner (since the same volume of sheath solution must now surround the larger-diameter liquid crystal core). What was also apparent was that the continuity of liquid crystal filling depended strongly on the flow rate. At low flow rates the fibers were not only thinner but the liquid crystal was also distributed in elongated drops along the fiber rather than forming a continuous thin core. As in the fibers with a non-chiral nematic mixture [12] the clearing point went up, the more so the thinner the fiber. In the thinnest the clearing point was about 15 K higher than in the bulk liquid crystal.
The selective reflection color was visible in most fibers, in case of the thickest fibers easily detectable even by the naked eye. Such mats had the blue-green color of the cholesteric mixture. On the other hand, in fibers produced with lower liquid crystal flow rate, thus with smaller inner diameter, the color could be strikingly different. In many cases it varied between blue-green and yellow but in certain locations it could even reach the red end of the spectrum, cf. Figure 7.13 showing two mats with slightly different fiber diameters. This modulation and quantization of the cholesteric selective reflection color are a consequence of the cylindrical boundary conditions imposed by the fiber sheath on the cholesteric helix.
Figure 7.13 The confinement of the cholesteric liquid crystal mixture 1 inside thin cylindrical fibers forces the director helix to expand or compress from its natural pitch, leading to a reflection color λ that depends on the inner fiber diameter d, as illustrated in the center diagram (black curve). The curve is calculated for a natural pitch p0 = 300 nm, average refractive index 1.5, and a director field as in Figure 7.14 (see reference [10] for details). As a guide for the eye the wavelengths of blue, green, and red have been indicated with horizontal lines in the respective color. The red color of the fiber shown in the left example is possible only for a fiber with inner diameter of about 400 nm. The blue-green color shown on the right could occur for even thinner fibers, but more likely is a somewhat greater inner fiber diameter of about 640 nm. The scale bar is 50 μm. (See the color version of this figure in Color Plates section.)
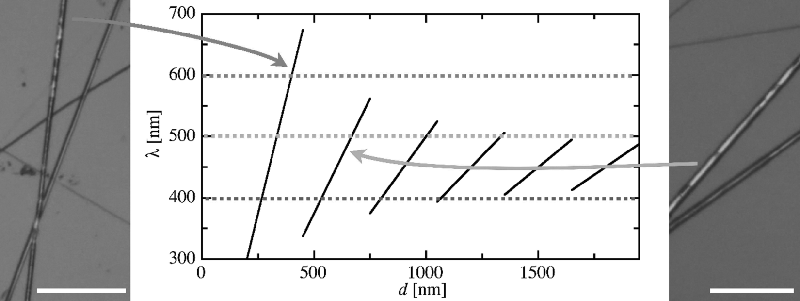
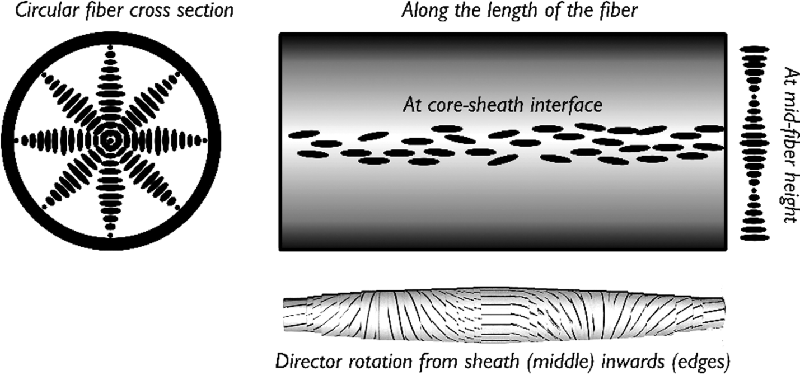
From the colored appearance of the fibers we can deduce that the director helix forms radially, compatible with a director aligned along the fiber at the boundary to the sheath. The geometry is illustrated in Figure 7.14. Note that this arrangement requires the director to be along the fiber axis also at the center of the fiber. This is the only central director orientation that is compatible with the cylindrical symmetry and allows a director distribution without a singular core (i.e., a disclination in which the core would be isotropic). In order to also fulfill the external boundary conditions, where the liquid crystal meets the polymer sheath, the inner diameter of the sheath must thus be a multiple of the helix pitch. Because we have a certain variation of fiber diameters we therefore expect a variation in color, as the pitch must adjust to the actual fiber diameter. The situation is similar to that of the classic experiment with a Cano wedge cell, but the cylindrical geometry is twice as restrictive on the helical pitch as the standard flat geometry. While the substrate distance in the Cano cell must be a multiple of half the pitch, the cylindrical fiber diameter must be an integer number times the full pitch, since the central boundary condition must also be fulfilled. This gives a strong quantization of color, to the extent that in a thin fiber it is the fiber inner diameter that effectively determines the color, cf. the diagram in Figure 7.13.
Figure 7.14 Schematic illustrations of the director field of a short-pitch cholesteric liquid crystal inside a cylindrical electrospun fiber.
In fibers with an inner diameter of about a micron or thicker the distortion away from the natural pitch will never be sufficient to account for the most extreme color changes, red reflection appearing from a cholesteric that is blue in its natural state. With a natural pitch p0 of 300 nm (greenish blue natural color, λ0 = 450 nm) we see in Figure 7.13 that red selective reflection would arise only if the inner fiber diameter is some 400 nm, allowing exactly one very extended helical pitch to fit in the fiber. This means that the fibers reflecting this color must in fact have an inner diameter of about this value. The majority of fibers are however more yellow-green, requiring an inner diameter that is either even thinner or somewhat thicker, around 650 nm. Having a most common inner diameter lower than some 400 nm in these fibers with micron-size outer diameter seems rather unlikely. It is thus possible to deduce from the colors that the fibers showing selective reflection have inner diameters in the range of 300–650 nm, most of them at the higher value. There were also several fibers in this study that did not show any visible selective reflection color. It is important to realize that this does not mean that they are unfilled or have the liquid crystal in a non-helical structure. The diagram in Figure 7.13 shows that the reflection wavelength of the cholesteric inside the fiber will lie in the UV range for many fiber diameters, hence no color can be seen from these fibers.
An important observation was that the transition between blue phase and cholesteric phase could no longer be detected for the liquid crystal in the fibers. This held for polarizing microscopy as well as DSC investigations, on heating as well as on cooling, and for all fibers regardless of their diameter. The reason is that the configuration in Figure 7.14 imposed by the cylindrical symmetry of the fiber, with a helical director twist in all directions perpendicular to the fiber axis, is unusual for a cholesteric phase in which the director normally twists only along a single direction. When confined inside the fiber the N* phase is thus forced to adopt a configuration that is in fact nothing else than the double-twist cylinder configuration that is the basic building block for blue phases (see model at the bottom of Figure 7.14). The same situation, albeit on much larger scale, was achieved by Heinz Kitzerow and co-workers [78] by filling cholesteric liquid crystals into glass capillaries.
Considering the blue phase-like director configuration imposed onto the N* phase by the cylindrical confinement, and the fact that the structural scale here is the same as that in bulk blue phases, it is easy to understand why the N*-BP* transition was not seen for mixture 1 in the fibers: the cylindrical confinement of the N* phase with strong surface director anchoring gives the N* phase a BP*-type structure, hence the two phases have become degenerate. For very thick fibers it should be possible to have a phase transition again, since multiple double-twist cylinders could then build up a cubic network in the BP* phase once the cylindrical confinement is less strong.
Fibers, filled with mixture 1 but with slightly sturdier sheath, achieved either by increasing the polymer concentration or by adding a small amount of the Ti(OiPr)4 sol–gel precursor to the outer spinning solution, were also investigated. Also these fibers exhibited selective reflection but continuous filling was never seen in these fibers. Instead the cholesteric was separated into small droplets spaced quite regularly along the fiber. The main reason for the different encapsulation of the liquid crystal here, compared to the earlier described fibers, is most likely the mismatch in viscosity of the sheath and core solutions. From experiments in the Xia group with coaxial electrospinning of mineral oil [64] and octadecane [25], respectively, inside a PVP + Ti(OiPr)4 sheath solution it is known that a change in viscosity of one of the fluids can have a dramatic impact on the continuity of the core. With the rather viscous mineral oil (viscosity about 70–80 mPa s) continuous and uniform filling of the fibers could easily be achieved with this sheath solution but in case of the much less viscous octadecane (heated to the liquid state prior to spinning, giving it a viscosity in the range of 2–4 mPa s) varicose break-up occurred inside the coaxial jet, resulting in regularly spaced drops of octadecane in the produced fibers.
7.5 Application Potential of Liquid Crystal-Containing Electrospun Fibers
The addition of the responsiveness and functionality of liquid crystals to the nano-to-microscale fibers produced by electrospinning produces a new material that clearly has strong potential for application. While much work so far has been done with hygroscopic polymers such as PVP, convenient for academic studies but generally problematic in a device context, a switch to textile-grade sheath material will open the door to employing the fibers in non-woven smart textiles. If a convenient way of applying electric fields to the fibers can be found, for instance by using carbon nanotube-containing fibers [79, 80], wearable displays might be realized in this way.
The strongly colored selective reflection from short-pitch cholesterics renders these phases most attractive and the temperature dependence of the pitch makes it straightforward to apply cholesterics as temperature sensors. Since we see the color also in fibers containing the cholesteric, autonomous (not needing any power supply) fiber mat-integrated sensors could easily be produced by electrospinning. One could for instance spin a web of cholesteric-containing fibers over an apparatus of arbitrary shape, giving it a surface that continuously reveals the temperature at every part, with full spatial resolution. Alternatively, pipes or tubes can be coated with the fibers to monitor the temperature of liquids or gases flowing through a complex system in a cheap and efficient way, useful for monitoring the performance of a cooling water system. Finally, with the right type of polymer sheath the cholesteric phase could be incorporated into synthetic textiles that could find use in clothing. Such textiles could obviously provide a temperature read-out, but if the fibers in addition are spun with a porous sheath and if the cholesteric mixture has an adequate composition, the textile could even function as a chemical sensor [81–83], useful for people working in hazardous environments.
Electrospun fibers from liquid crystalline elastomers have obvious application potential in actuators. Judging from the lack of information on the actuation properties of the fibers produced so far [15] there are still problems to be addressed, but the concept is certainly very attractive and should be further explored. Many liquid crystal types have not yet been integrated into electrospun fibers, e.g., ferroelectric and discotic liquid crystals. They would allow the production of new functional fibers for instance with spontaneous electric polarization or photoconductivity, opening for various new application possibilities, for instance in energy conversion [84–87]. The future looks bright for liquid crystal electrospinning and we can expect to see some very exciting developments in this field in the coming years.
1. S. Agarwal and A. Greiner. Nanofibers by electrospinning. Polymers Adv. Tech. 2011, 22, 293–294.
2. X. Lu, C. Wang, and Y. Wei. One-dimensional composite nanomaterials: synthesis by electrospinning and their applications. Small 2009, 5, 2349–2370.
3. D. Reneker and A. Yarin. Electrospinning jets and polymer nanofibers. Polymer 2008, 49, 2387–2425.
4. J. Schiffman and C. Schauer. A review: electrospinning of biopolymer nanofibers and their applications. Polymer Rev. 2008, 48, 317–352.
5. G. Rutledge and S. Fridrikh. Formation of fibers by electrospinning. Adv. Drug Deliv. Rev. 2007, 59, 1384–1391.
6. A. Greiner and J. Wendorff. Electrospinning: a fascinating method for the preparation of ultrathin fibres. Angew. Chem. Int. Ed. 2007, 46, 5670–5703.
7. D. Li and Y. Xia. Electrospinning of nanofibers: reinventing the wheel? Adv. Mater. 2004, 16, 1151–1170.
8. Z. Huang, Y. Zhang, M. Kotaki, and S. Ramakrishna. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Composites Sci. Tech. 2003, 63, 2223–2253.
9. E. A. Buyuktanir, W. Frey, M. W. Margaret, and J. L. West. Self-assembled, optically responsive nematic liquid crystal/polymer core–shell fibers: formation and characterization. Polymer 2010, 51, 4823–4830.
10. E. Enz and J. Lagerwall. Electrospun microfibres with temperature sensitive iridescence from encapsulated cholesteric liquid crystal. J. Mater. Chem. 2010, 20, 6866–6872.
11. E. Enz, U. Baumeister, and J. Lagerwall. Coaxial electrospinning of liquid crystal-containing poly(vinyl pyrrolidone) microfibers. Beilstein J. Org. Chem. 2009, 5, doi:
12. J. P. F. Lagerwall, J. T. McCann, E. Formo, G. Scalia, and Y. Xia. Coaxial electrospinning of microfibres with liquid crystal in the core. Chem. Commun. 2008, 5420–5422.
13. Y. Wu, Q. An, J. Yin, T. Hua, H. Xie, G. Li, and H. Tang. Liquid crystal fibers produced by using electrospinning technique. Colloid Polymer Sci. 2008, 286, 897–905.
14. J. Canejo, J. Borges, M. Godinho, P. Brogueira, P. Teixeria, and E. Terentjev. Helical twisting of electrospun liquid crystalline cellulose micro-and nanofibers. Adv. Mater. 2008, 20, 4821–4825.
15. S. Krause, R. Dersch, J. Wendorff, and H. Finkelmann. Photocrosslinkable liquid crystal main-chain polymers: thin films and electrospinning. Macromol. Rapid Commun. 2007, 28, 2062–2068.
16. J. F. Cooley. US Patent 692,631 (1902).
17. W. J. Morton. US Patent 705,691 (1902).
18. A. Formhals.Process and apparatus for preparing artificial threads. US Patent 1,975,504 (A) (1934).
19. J. Yu, S. Fridrikh, and G. Rutledge. The role of elasticity in the formation of electrospun fibers. Polymer 2006, 47, 4789–4797.
20. K. Arayanarakul, N. Choktaweesap, D. Aht-ong, C. Meechaisue, and P. Supaphol. Effects of poly(ethylene glycol), inorganic salt, sodium dodecyl sulfate, and solvent system on electrospinning of poly(ethylene oxide). Macromol. Mater. Eng. 2006, 291, 581–591.
21. S. Bhattarai, N. Bhattarai, H. Yi, P. Hwang, D. Cha, and H. Kim. Novel biodegradable electrospun membrane: scaffold for tissue engineering. Biomaterials 2004, 25, 2595–2602.
22. D. Li, Y. Wang, and Y. Xia. Electrospinning of polymeric and ceramic nanofibers as uniaxially aligned arrays. Nano Lett. 2003, 3, 1167–1171.
23. D. Li, Y. Wang, and Y. Xia. Electrospinning nanofibers as uniaxially aligned arrays and layer-by-layer stacked films. Adv. Mater. 2004, 16, 361–366.
24. J. McCann, D. Li, and Y. Xia. Electrospinning of nanofibers with core–sheath, hollow, or porous structures. J. Mater. Chem. 2005, 15, 735–738.
25. J. McCann, M. Marquez, and Y. Xia. Melt coaxial electrospinning: a versatile method for the encapsulation of solid materials and fabrication of phase change nanofibers. Nano Lett. 2006, 6, 2868–2872.
26. W. Salalha, J. Kuhn, Y. Dror, and E. Zussman. Encapsulation of bacteria and viruses in electrospun nanofibres. Nanotechnology 2006, 17, 4675–4681.
27. C. Zhang, X. Yuan, L. Wu, Y. Han, and J. Sheng. Study on morphology of electrospun poly(vinyl alcohol) mats. Eur. Polym. J. 2005, 41, 423–432.
28. Z. Jun, H. Hou, J. Wendorff, and A. Greiner. Poly(vinyl alcohol) nanofibres by electrospinning: influence of molecular weight on fibre shape. E-polymers 2005, 038.
29. N. Li, X. Qin, E. Yang, and S. Wang. Effect on instability section of PVA electrospinning nanofibers by adding LiCl. Mater. Lett. 2008, 62, 1345–1348.
30. N. Li, X. Qin, L. Lin, and S. Wang. The effects of spinning conditions on the morphology of electrospun jet and nonwoven membrane. Polymer Eng. Sci. 2008, 48, 2362–2366.
31. D. He, B. Hu, Q. Yao, K. Wang, and S. Yu. Large-scale synthesis of flexible freestanding SERS substrates with high sensitivity: electrospun PVA nanofibers embedded with controlled alignment of silver nanoparticles. ACS Nano 2009, 3, 3993–4002.
32. H. Sun, H. Zhang, J. Zhang, H. Wei, J. Ju, M. Li, and B. Yang. White-light emission nanofibers obtained from assembling aqueous single-colored CdTe NCs into a PPV precursor and PVA matrix. J. Mater. Chem. 2009, 19, 6740–6744.
33. B. Sundaray, V. Subramanian, T. Natarajan, R. Xiang, C. Chang, and W. Fann. Electrospinning of continuous aligned polymer fibers. Appl. Phys. Lett. 2004, 84, 1222–1224.
34. S. Megelski, J. Stephens, D. Chase, and J. Rabolt. Micro- and nanostructured surface morphology on electrospun polymer fibers. Macromolecules 2002, 35, 8456–8466.
35. R. Sen, B. Zhao, D. Perea, M. Itkis, H. Hu, J. Love, E. Bekyarova, and R. Haddon. Preparation of single-walled carbon nanotube reinforced polystyrene and polyurethane nanofibers and membranes by electrospinning. Nano Lett. 2004, 4, 459–464.
36. C. Casper, J. Stephens, N. Tassi, D. Chase, and J. Rabolt. Controlling surface morphology of electrospun polystyrene fibers: effect of humidity and molecular weight in the electrospinning process. Macromolecules 2004, 37, 573–578.
37. S. Wang, Y. Li, J. Bai, Q. Yang, Y. Song, and C. Zhang. Characterization and photoluminescence studies of CdTe nanoparticles before and after transfer from liquid phase to polystyrene. Bull. Mater. Sci. 2009, 32, 487–491.
38. G. Eda and S. Shivkumar. Bead-to-fiber transition in electrospun polystyrene. J. Appl. Polymer Sci. 2007, 106, 475–487.
39. G. Eda, J. Liu, and S. Shivkumar. Flight path of electrospun polystyrene solutions: effects of molecular weight and concentration. Mater. Lett. 2007, 61, 1451–1455.
40. R. Krishnappa, K. Desai, and C. Sung. Morphological study of electrospun polycarbonates as a function of the solvent and processing voltage. J. Mater. Sci. 2003, 38, 2357–2365.
41. G. Kim, G. Michler, and P. Potschke. Deformation processes of ultrahigh porous multi-walled carbon nanotubes/polycarbonate composite fibers prepared by electrospinning. Polymer 2005, 46, 7346–7351.
42. S. Moon and R. Farris. The morphology, mechanical properties, and flammability of aligned electrospun polycarbonate (PC) nanofibers. Polymer Eng. Sci. 2008, 48, 1848–1854.
43. J. McCann, M. Marquez, and Y. Xia. Highly porous fibers by electrospinning into a cryogenic liquid. J. Am. Chem. Soc. 2006, 128, 1436–1437.
44. J. Liu, Z. Yue, and H. Fong. Continuous nanoscale carbon fibers with superior mechanical strength. Small 2009, 5, 536–542.
45. E. J. Ra, T. H. Kim, W. J. Yu, K. H. An, and Y. H. Lee. Ultramicropore formation in PAN/camphor-based carbon nanofiber paper. Chem. Commun. 2010, 46, 1320–1322.
46. C. Lai, Q. Guo, X. Wu, D. Reneker, and H. Hou. Growth of carbon nanostructures on carbonized electrospun nanofibers with palladium nanoparticles. Nanotechnology 2008, 19, 195303.
47. Q. Zhang, Z. Chang, M. Zhu, X. Mo, and D. Chen. Electrospun carbon nanotube composite nanofibres with uniaxially aligned arrays. Nanotechnology 2007, 18, 115611.
48. A. Bianco, G. Iardino, A. Manuelli, C. Bertarelli, and G. Zerbi. Strong orientation of polymer chains and small photochromic molecules in polyamide 6 electrospun fibers. ChemPhysChem 2007, 8, 510–514.
49. C. Mit-uppatham, M. Nithitanakul, and P. Supaphol. Ultrafine electrospun polyamide-6 fibers: effect of solution conditions on morphology and average fiber diameter. Macromol. Chem. Phys. 2004, 205, 2327–2338.
50. R. Dersch, T. Liu, A. Schaper, A. Greiner, and J. Wendorff. Electrospun nanofibers: internal structure and intrinsic orientation. J. Polymer Sci. Part A: Polymer Chem. 2003, 41, 545–553.
51. Y. Yao, Z. Gu, J. Zhang, C. Pan, Y. Zhang, and H. Wei. Fiber-oriented liquid crystal polarizers based on anisotropic electrospinning. Adv. Mater. 2007, 19, 3707.
52. X. Sui and H. Wagner. Tough nanocomposites: the role of carbon nanotube type. Nano Lett. 2009, 9, 1423–1426.
53. A. Bazilevsky, A. Yarin, and C. Megaridis. Co-electrospinning of core–shell fibers using a single-nozzle technique. Langmuir 2007, 23, 2311–2314.
54. P. L. Almeida, S. Kundu, J. P. Borges, M. H. Godinho, and J. L. Figueirinhas. Electrooptical light scattering shutter using electrospun cellulose-based nano-and microfibers. Appl. Phys. Lett. 2009, 95, 043501.
55. M. Acharya, G. Arumugam, and P. Heiden. Dual electric field induced alignment of electrospun nanofibers. Macromol. Mater. Eng. 2008, 293, 666–674.
56. D. Reneker and I. Chun. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology 1996, 7, 216–223.
57. D. Reneker, A. Yarin, H. Fong, and S. Koombhongse. Bending instability of electrically charged liquid jets of polymer solutions in electrospinning. J. Appl. Phys. 2000, 87, 4531–4547.
58. P. Katta, M. Alessandro, R. Ramsier, and G. Chase. Continuous electrospinning of aligned polymer nanofibers onto a wire drum collector. Nano Lett. 2004, 4, 2215–2218.
59. L. Carnell, E. Siochi, N. Holloway, R. Stephens, C. Rhim, L. Niklason, and R. Clark. Aligned mats from electrospun single fibers. Macromolecules 2008, 41, 5345–5349.
60. H. Yan, L. Liu, and Z. Zhang. Alignment of electrospun nanofibers using dielectric materials. Appl. Phys. Lett. 2009, 95, 143114.
61. C. Hellmann, J. Belardi, R. Dersch, A. Greiner, J. Wendorff, and S. Bahnmueller. High precision deposition electrospinning of nanofibers and nanofiber nonwovens. Polymer 2009, 50, 1197–1205.
62. C. Chang, K. Limkrailassiri, and L. Lin. Continuous near-field electrospinning for large area deposition of orderly nanofiber patterns. Appl. Phys. Lett. 2008, 93, 123111.
63. D. Sun, C. Chang, S. Li, and L. Lin. Near-field electrospinning. Nano Lett. 2006, 6, 839–842.
64. D. Li and Y. Xia. Direct fabrication of composite and ceramic hollow nanofibers by electrospinning. Nano Lett. 2004, 4, 933–938.
65. Z. Sun, E. Zussman, A. Yarin, J. Wendorff, and A. Greiner. Compound core–shell polymer nanofibers by co-electrospinning. Adv. Mater. 2003, 15, 1929.
66. K. Nakashima, K. Tsuboi, H. Matsumoto, R. Ishige, M. Tokita, J. Watanabe, and A. Tanioka. Control over internal structure of liquid crystal polymer nanofibers by electrospinning. Macromol. Rapid Commun. 2010, 31, 1641–1645.
67. N. Ogata, N. Shimada, S. Yamaguchi, K. Nakane, and T. Ogihara. Meltelectrospinning of poly(ethylene terephthalate) and polyalirate. J. Appl. Polymer Sci. 2007, 105, 1127–1132.
68. W. Pisula, M. Zorn, J. Y. Chang, K. Muellen, and R. Zentel. Liquid crystalline ordering and charge transport in semiconducting materials. Macromol. Rapid Commun. 2009, 30, 1179–1202.
69. Y. Wu, Q. An, J. Yin, T. Hua, H. Xie, G. Li, and H. Tang. Liquid crystal fibers produced by using electrospinning technique. Colloid Polymer Sci. 2008, 286, 897–905.
70. P. Rudquist, J. P. F. Lagerwall, M. Buivydas, F. Gouda, S. T. Lagerwall, N. A. Clark, J. E. Maclennan, R. F. Shao, D. Coleman, S. Bardon, T. Bellini, D. R. Link, G. Natale, M. A. Glaser, D. M. Walba, M. D. Wand, and X. H. Chen. The case of threshold-less antiferroelectricity: polarization-stabilized twisted SmC* liquid crystals give V-shaped electro-optic response. J. Mater. Chem. 1999, 9, 1257–1261.
71. G. Iannacchione, G. Crawford, S. Qian, J. Doane, and D. Finotello. Nematic ordering in highly restrictive Vycor glass. Phys. Rev. E 1996, 53, 2402–2411.
72. J. P. F. Lagerwall, D. D. Parghi, D. Kruerke, F. Gouda, and P. Jagemalm. Phases, phase transitions and confinement effects in a series of antiferroelectric liquid crystals. Liq. Cryst. 2002, 29, 163–178.
73. R. Leheny, S. Park, R. Birgeneau, J. Gallani, C. Garland, and G. Iannacchione. Smectic ordering in liquid-crystal–aerosil dispersions. I. X-ray scattering. Phys. Rev. E 2003, 67, 011708.
74. T. Jin, B. Zalar, A. Lebar, M. Vilfan, S. Zumer, and D. Finotello. Anchoring and structural transitions as a function of molecular length in confined liquid crystals. Eur. Phys. J. E 2005, 16, 159–165.
75. M. Steinhart, S. Zimmermann, P. Goring, A. Schaper, U. Gosele, C. Weder, and J. Wendorff. Liquid crystalline nanowires in porous alumina: geometric confinement versus influence of pore walls. Nano Lett. 2005, 5, 429–434.
76. G. Carbone, R. Barberi, I. Musevic, and U. Krzic. Atomic force microscope study of presmectic modulation in the nematic and isotropic phases of the liquid crystal octylcyanobiphenyl using piezoresistive force detection. Phys. Rev. E 2005, 71, 051704.
77. K. Kocevar and I. Musevic. AFM study of forces in and structures of nematic liquid crystal interfaces on silanated glass. Liq. Cryst. 2001, 28, 599–606.
78. H. Kitzerow, B. Liu, F. Xu, and P. Crooker. Effect of chirality on liquid crystals in capillary tubes with parallel and perpendicular anchoring. Phys. Rev. E 1996, 54, 568–575.
79. L. Hu, M. Pasta, F. Mantia, L. Cui, S. Jeong, H. Deshazer, J. Choi, S. Han, and Y. Cui. Stretchable, porous, and conductive energy textiles. Nano Lett. 2010, 10, 708–714.
80. B. Shim, W. Chen, C. Doty, C. Xu, and N. Kotov. Smart electronic yarns and wearable fabrics for human biomonitoring made by carbon nanotube coating with polyelectrolytes. Nano Lett. 2008, 8, 4151–4157.
81. Y. Han, K. Pacheco, C. W. M. Bastiaansen, D. J. Broer, and R. P. Sijbesma. Optical monitoring of gases with cholesteric liquid crystals. J. Am. Chem. Soc. 2010, 132, 2961–2967.
82. D. Winterbottom, R. Narayanaswamy, and I. Raimundo. Cholesteric liquid crystals for detection of organic vapours. Sen. Actuators B 2003, 90, 52–57.
83. F. Dickert, A. Haunschild, and P. Hofmann. Cholesteric liquid-crystals for solvent vapor detection-elimination of cross-sensitivity by band shape-analysis and pattern-recognition. Fresen. J. Anal. Chem. 1994, 350, 577–581.
84. A. Jakli. Electro-mechanical effects in liquid crystals. Liq. Cryst. 2010, 37, 825–837.
85. J. Harden, M. Chambers, R. Verduzco, P. Luchette, J. T. Gleeson, S. Sprunt, and A. Jakli. Giant flexoelectricity in bent-core nematic liquid crystal elastomers. Appl. Phys. Lett. 2010, 96, 102907.
86. M. Chambers, R. Verduzco, J. T. Gleeson, S. Sprunt, and A. Jakli. Flexoelectricity of a calamitic liquid crystal elastomer swollen with a bent-core liquid crystal. J. Mater. Chem. 2009, 19, 7909–7913.
87. M. Chambers, R. Verduzco, J. T. Gleeson, S. Sprunt, and A. Jakli. Calamitic liquid-crystalline elastomers swollen in bent-core liquid-crystal solvents. Adv. Mater. 2009, 21, 1622.