
Chapter 8
Functional Liquid Crystalline Block Copolymers: Order Meets Self-Assembled Nanostructures
8.1 What are Functional Liquid Crystalline Block Copolymers?
Liquid crystalline block copolymers (LC-BCPs) are block copolymers of which one or more constituting polymers have mesogens either as side groups or in the main chain. The great interest of LC-BCPs has long been recognized. The most important feature is the interplay between two levels of order and self-assembly: the LC order formed by mesogens and the ordered nanostructures arising from the microphase separation of the blocks. There have been many studies on the mutual influence of the two levels of order in LC-BCPs [1–10]. A representative example was reported by Finkelmann and co-workers in 2000 [8]. It was found that when the microphase-separation occurred in the isotropic phase of a LC-BCP, the gyroid morphology was formed, while when the microphase separation was allowed to develop in a smectic phase, observed was a lamellar morphology. It is easy to picture that a layered smectic order of mesogens accommodates well a lamellar structure and thus can promote its formation.
In a broad sense, all LC-BCPs are functional and entitled with a combination of the characteristic properties of liquid crystalline polymers (LCPs) and BCPs: they can form LC phases and give rise to a variety of self-assembled nanostructures. However, what we mean by functional LC-BCPs in this chapter is different. A number of studies on LC-BCPs in recent years have explored the interplay of LC order and self-assembled nanostructures with the purpose to generate a specific new function. In other words, by rational LC-BCP design, an “active” interaction between two levels of order can be used to achieve something that otherwise is unachievable, rather than an effect of a “passive” interaction, which often is the consequence of the coexisting LC order and nanostructures. We focus our review and discussion only on representative studies that demonstrated a specific function as a result of the interaction between LC order and a self-assembled nanostructure.
8.2 Macroscopic Orientation of Nanodomains
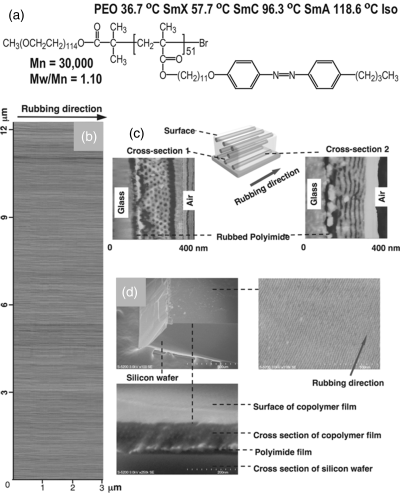
The most striking demonstration of new functions of LC-BCPs is the possibility of using the orientation of mesogens (LC order) on one block to align the nanodomains (nanocylinders or stripe-like domains) of another block at a macroscopic scale [11, 12]. In a study by Yu et al. [13], a thin film (~200 nm thick) of a diblock copolymer composed of poly(ethylene oxide) (PEO) and a polymethacrylate bearing azobenzene mesogenic side groups was spin-coated onto a glass slide coated with a rubbed polyimide layer; on slow cooling from the isotropic state into the smectic phases, surface-induced orientation of mesogens could bring microphase separated PEO cylindrical nanodomains (nanocylinders) to align in the same direction as mesogens. At room temperature, uniformly oriented crystalline PEO nanocylinders were found in an area of 12.4 × 3.1 μm by AFM imaging. An example of the reported results is shown in Figure 8.1, together with the chemical structure and phase transition temperatures of the used LC-BCP. This study shows clearly that the surface-induced orientation of the mesogens (a typical LC orientation method) builds an anisotropic matrix that imposes the orientation of PEO nanodomains. By this mechanism, the orientation of nanodomains can extend to the whole area of rubbed surface. This approach provides a valuable new means to control the order and organization of self-assembled nanostructures in BCPs, which often is a key requirement for enabling their applications [14, 15].
Figure 8.1 (a) Chemical structure and phase transition temperatures of the LC diblock copolymer. (b) Macroscopic orientation of PEO nanocylinders along the rubbing direction in an area of 12.4 × 3.1 μm. (c) AFM phase images. (d) SEM images of the cross sections of the film (the bright lines and dots in the SEM images are PEO blocks selectively stained by RuO4). Source: Yu et al. (2006). Reprinted with permission. (See the color version of this figure in Color Plates section.)
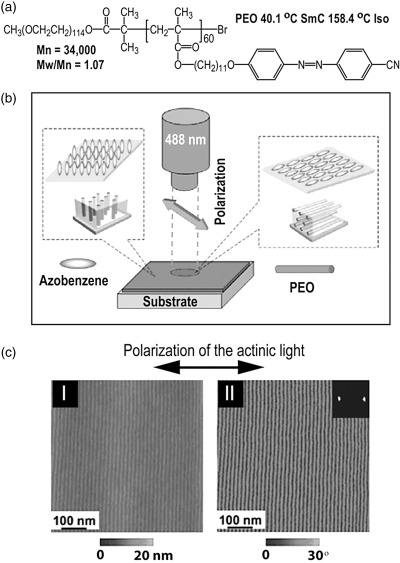
With azobenzene as the mesogens, the long-range LC orientation can be induced optically as a result of the trans–cis photoisomerization of the chromophore [16]. Yu et al. then used a similar LC-BCP and demonstrated that a macroscopic orientation of PEO nanodomains could also be obtained using the photo-induced LC orientation [17]. In this case, a film of about 100-nm thick, cast onto a non-rubbed glass slide, was exposed to linearly polarized visible light (488 nm, 100 mW/cm2) at room temperature and then annealed at 140°C, yielding uniform, in-plane orientation of PEO nanocylinders in the same direction as azobenzene mesogens, that is, perpendicular to the polarization of the visible light, as shown in Figure 8.2. Using photo-induced LC orientation to align PEO nanodomains, the thermal annealing, which is required to enhance the LC orientation, should be carried out at a temperature where PEO is in the melted, fluid state in order to facilitate the orientation of the nanodomains under the effect of the LC matrix. In principle, the macroscopic orientation of PEO nanocylinders could be obtained in a large area whose size is basically determined by the light exposure process and conditions.
Figure 8.2 (a) Chemical structure and phase transition temperatures of the used LC diblock copolymer. (b) Schematic illustration of the orientation of azobenzene mesogens and the resulting alignment of microphase-separated nanocylinders in the irradiated and non-irradiated area of the block copolymer films. (c) AFM images of a film in an irradiated area: topological (I) and phase image (II). Source: Yu et al. (2006). Reprinted with permission.
The use of light to direct the orientation of mesogens and thus the orientation of the nanodomains in LC-BCPs is not just about an orientation technique differing from surface rubbing, it offers appealing advantages associated with light. The most interesting may be the easy spatial control because the area of a thin film exposed to light can be chosen by using either a photomask or an interference pattern. Seki et al. exploited this possibility [18, 19]. Using a diblock copolymer composed of polystyrene (PS) and an azobenzene LCP, they exposed a thin film (thickness ~100 nm) containing vertically aligned PS nanocylinders to linearly polarized light through a photomask (10-μm period), and found alternating regions with PS nanocylinders oriented in-plane (in exposed regions) and perpendicularly to the substrate surface (in non-exposed regions) could be patterned, with a sharp transition of the orientational state at the interface [18]. An example of the results is shown in Figure 8.3. In principle, the photo-induced LC orientation could be explored to pattern or organize the orientation of nanodomains in LC-BCPs in a three-dimensional (3D) fashion.
Figure 8.3 (a) Chemical structure and phase transition temperatures of the used LC diblock copolymer. (b) AFM phase image of the initial film showing that polystyrene nanocylinders are aligned perpendicularly to the substrate surface. (c) AFM topographical image after patterned irradiation with linearly polarized light. (d–f) AFM phase images recorded at irradiated area (d), boundary region (e) and non-irradiated area (f). Source: Morikawa et al. (2007). Reprinted with permission.

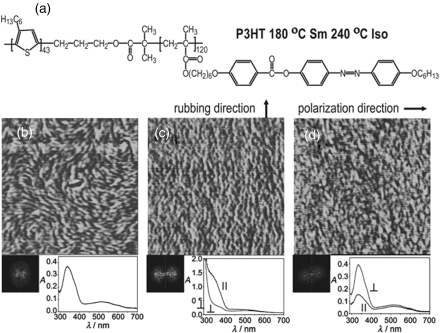
Recently, our group extended the approach to π-conjugated polymers by demonstrating the use of LC orientation to obtain macroscopically aligned nanodomains of poly(3-hexylthiophene) (P3HT), which is one of the most studied conducting polymers. There is growing interest in BCPs comprising one or more conducting polymers, because the microphase separation in BCPs provides a simple and effective way to generate a microstructure or morphology useful for their electronic and optoelectronic applications [20–22]. Macroscopically aligning self-assembled nanodomains of regioregular P3HT is challenging because it has a very high melting temperature Tm (>200oC), which means that its crystallization is the dominant event and competes with the self-assembly or an organization process [21]. We rationally designed and synthesized a diblock copolymer composed of P3HT and an azobenzene LCP of that the clearing (LC-to-isotropic transition) temperature is greater than the Tm of P3HT [23]. Under this condition, it is possible to anneal a thin film of the LC-BCP in a LC phase to obtain surface-induced orientation of mesogens, while retaining P3HT in its melted state to allow the orientation of fluid P3HT nanodomains inside the oriented LC matrix. The results in Figure 8.4 shows that both surface and photo-induced orientation of azobenzene mesogens in the major phase of the LC-BCP could be used to impose a macroscopic orientation of stripe-like nanodomains of P3HT in the same direction as the mesogens. In the case of using surface-induced LC orientation, a thin film (~30 nm thick) cast on a glass slide with rubbed surface was first cooled from 260°C (isotropic state for both blocks) to 220°C (LC phase for the LCP block but isotropic phase for P3HT) for the orientation of both azobenzene mesogens and microphase separated P3HT strip-like domains, and then cooled to room temperature for the crystallization of P3HT. For photo-induced LC orientation, it was conducted at 45°C, and, prior to the analysis at room temperature, the film was annealed at 120°C. In view of the great ease of LC orientation and the variety of LC orientation methods, this study demonstrated a promising means for achieving and manipulating macroscopically ordered microstructure or morphology of π-conjugated polymer-based BCPs. As a future development, it would be interesting, and challenging, to know how to use this approach to induce macroscopic orientation of the nanodomains of both a donor and an acceptor polymer, which is required for photovoltaic applications. One possible strategy is to develop donor–acceptor diblock copolymers of which the two blocks are LCP.
Figure 8.4 (a) Chemical structure and phase transition temperatures of the LC diblock copolymer. (b–d) AFM phase images (1×1 μm), fast Fourier transformations and polarized absorption spectra for thin films cast on a nonrubbed quartz plate (b), a rubbed glass plate for surface-induced orientation of the mesogens (c), and a nonrubbed quartz plate subjected to irradiation for photoinduced orientation of the mesogens (d). Absorption spectra were recorded with the beam of the spectrophotometer polarized to be parallel and perpendicular, respectively, to either the surface rubbing direction (c) or the polarization of the irradiating light (d). The surface- or photo-induced orientation of azobenzene mesogens can be noticed from the dichroism of the absorption band of trans azobenzene around 360 nm, while the absorption band near 540 nm is from crystallized P3HT chains. Source: Han et al. (2010). Reprinted with permission. (See the color version of this figure in Color Plates section.)
8.3 Shape-Memory Materials and Elastomers
LC thermoplastic elastomer represents another case where the interplay of LC order and a self-assembled BCP nanostructure can generate new functions. A thermoplastic elastomer can be obtained with an ABA-type triblock copolymer if the central B block is rubbery with a low Tg while the end A blocks are crystalline (or amorphous) with a high Tm (or Tg). The microphase separation could result in a network structure with the rubbery B blocks interconnected by rigid nanodomains of the A blocks; upon elongation, the extension (orientation) of B chains can be supported by the nanodomains of A acting as physical cross-links. The importance of this nanostructure has long been known and led to the development of commercialized thermoplastic elastomers like SBS (styrene-b-butadiene-b-styrene) or SIS (styrene-b-isoprene-b-styrene). Now, if one of the constituting polymers is LCP, the elastic deformation makes it easy to induce orientation of mesogens, and this could give rise to fascinating materials.
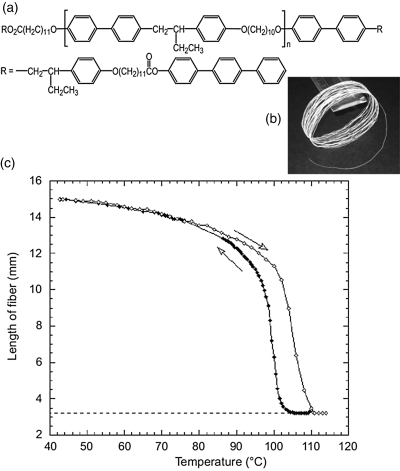
Such an example can be found in a study by Terentjev et al. [24]. They prepared a triblock copolymer whose middle block is a main-chain nematic LCP with a Tg at ~40°C and whose end blocks are a short segment containing a semi-crystalline terphenyl moiety. With this LC-BCP, fiber could readily be drawn from the isotropic state; while upon subsequent cooling into the nematic phase, a uniform orientation of mesogens (monodomain) and the microphase separation was found to take place simultaneously, resulting in a network structure in which aggregated terphenyl moieties could act as multifunctional junction points and retain the orientation of mesogens. With mesogens incorporated into the main chain, the conformation of the chain backbone is strongly coupled with the ordering state of the mesogens. The resulting fiber exhibits a striking shape-memory effect. As shown in Figure 8.5, when the fiber is heated to above the clearing (nematic–isotropic) temperature, the order–disorder phase transition of the mesogens induces a conformational transition of chain backbone from an extended to a random coil state, the macroscopic manifestation of which is a nearly 500 percent contraction of the fiber. On cooling, the formation of the nematic phase restores the uniform orientation of mesogens and the fiber recovers its initial length. Since the reversible LC phase transition is responsible for the shape change, the large contraction and elongation of the fiber upon heating and cooling are reversible. This feature is unique to LC-BCPs, which contracts with conventional shape-memory polymers for which the temporary to permanent shape recovery occurs only once without reset of the temporary shape [25, 26].
Figure 8.5 (a) Chemical structure of the used LC (telechelic) triblock copolymer. (b) Picture showing a roll of drawn fiber of the polymer. (c) Change in the length of a freely suspended fiber during a heating and cooling cycle, showing the reversible shape-memory effect. Source: Ahir et al. (2006). Reprinted with permission.
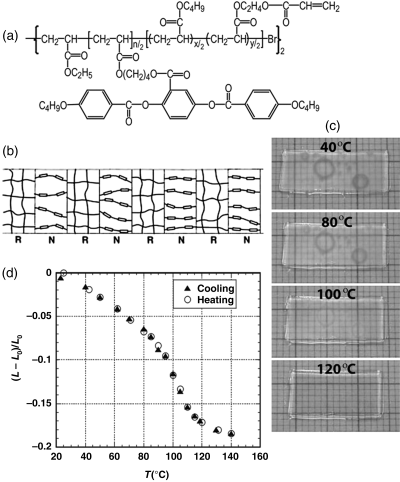
Li et al. exploited this reversible shape-memory effect in a different way by targeting the fabrication of an artificial muscle [27]. Instead of using a main-chain nematic polymer, they used a side-on LCP that also exhibits a strong coupling between the LC order and the chain backbone [28]. The advantage of this design resides in the fact that controlled radical polymerization techniques such as atom transfer radical polymerization (ATRP) could be utilized for the synthesis of the LC-BCP with a low polydispersity. Their triblock copolymer consisted in a side-on LCP middle block and two rubbery end blocks (a random copolymer of n-butyl acrylate and an acrylate comonomer bearing a photo-polymerizable unit); by having an appropriate weight ratio of nematic to rubbery blocks (3:2), a lamellar structure was obtained with alternating LCP and rubbery nanodomains. LC elastomers with such a striated structure were proposed by De Gennes to behave like artificial muscle [29]. Li et al. used a magnetic field to induce a uniform orientation of mesogens in the LCP nanodomains while cross-lining the rubbery blocks, and observed a reversible contraction and elongation of a thick film (0.16 mm thickness) upon thermal LC-isotropic phase transition, as shown in Figure 8.6.
Figure 8.6 (a) Chemical structure of the used LC triblock copolymer. (b) A model of striated artificial muscles based on a microphase separated lamellar structure (N and R denote nematic polymer and cross-linked elastomer nanodomains, respectively). (c) Pictures of a sample taken at different temperatures showing a contraction upon the nematic-isotropic phase transition (near 110°C). (d) Fractional change in the length of the film during a heating and cooling cycle (L0 and L are the initial and the changing lengths of the film, respectively). Source: Li et al. (2004). Reprinted with permission.
In addition to the use of LC triblock copolymers as shape-memory materials, they can also be designed to accomplish other functions. Kornfield et al. designed a triblock copolymer with polystyrene end blocks and a very long LC middle block [30]. When added in a small-molecule nematic LC, the BCP self-assemble into a network structure with the LCP block dissolved by the nematic solvent and PS chains microphase separated into micellar nanodomains acting as cross-linking points. Such LC gels were shown to display excellent electro-optical properties. In a study carried out by our group, a LCP with azobenzene mesogens was grafted onto the rubbery polybutadiene block of SBS [31, 32]. The resulting thermoplastic elastomer, whose films can easily be prepared by solution-casting, could be used to record mechanically tunable diffraction gratings. Typically, when a film under strain with a uniform orientation of mesogens, is exposed to UV light through a grating photomask, a diffraction grating is formed as a result of the erasure of LC orientation in exposed areas. We showed that the period, diffraction efficiency and diffraction mode (between Raman-Nath and Bragg regimes) of such an elastic grating could be reversibly tuned upon elastic elongation or contraction of the film.
In all the above examples, a LCP is the active middle block, while it can also be used as the end blocks. We designed and synthesized a thermoplastic elastomer comprising poly(n-butyl acrylate) (PnBA) as the rubbery middle block and a LCP bearing azobenzene mesogens as the end blocks [33]. In this case, as the physical cross-links were made with a LCP, new features emerged. When a film of this LC-BCP is stretched at T < Tg of the LCP end blocks, the polymer acts as a conventional thermoplastic elastomer, with the elastic extension of PnBA chains supported by glassy cylindrical nanodomains of the LCP in which there is no long-range molecular orientation of mesogens. Interestingly, this elastomer can exhibit an intermediate elastic regime. As schematically illustrated in Figure 8.7, when a film is stretched with the LCP in its nematic phase, that is, above Tg but below the nematic–isotropic phase transition, the degree of elastic deformation of PnBA chains is reduced due to a deformation of the LCP nanodomains that induces a long-range orientation of mesogens. This LC orientation can be retained in the relaxed film at room temperature, giving rise to a thermoplastic elastomer containing glassy microdomains with oriented azobenzene mesogens. Subsequent deformation from this thermoplastic elastomer with anisotropic physical cross-links is reversible.
Figure 8.7 (a) Chemical structure and phase transition temperatures of the LC triblock copolymer. (b) Schematic illustration of the elasticity and orientation states in the thermoplastic elastomer: (I) initial film before stretching, showing no long-range orientation of mesogens in the microphase separated azo polymer nanodomains; (II) stretching at T < Tg of the azo polymer (glassy microdomains), showing the conventional elasticity; (III) stretching at Tni > T > Tg of the azo polymer (LC microdomains), showing an intermediate elastomer state with deformed azo polymer nanodomains and induced LC orientation; (IV) relaxation at T < Tg (glassy microdomains with oriented mesogens). The elastic recovery of the film is indicated by arrows. Source: Cui et al. (2004). Reprinted with permission.

8.4 Stimuli-Responsive Vesicles in Solution
LC order can also meet with self-assembled nanostructures in a solution. In principle, any LC-BCPs dissolved in a block-selective solvent could self-assemble into micelles or vesicles. Of particular interest are amphiphilic LC-BCPs built with a hydrophobic LCP and a hydrophilic polymer. They tend to form large vesicles with the LCP constituting the membrane (wall of the capsule) [34, 35]. This observation itself reflects the influence of the LC order on the self-assembled nanostructure as smaller curvature in a large membrane is more amenable to long-range order of mesogens than a spherical core of a core–shell micelle. BCP vesicles have attracted much attention over the last two decades or so as they could potentially be used as nanovectors for controlled drug delivery applications [36, 37]. Typically, a water-soluble agent can be loaded inside a BCP vesicle in aqueous solution, with the hydrophobic membrane providing a good protection. At the time of release (after the vesicle reaches a target site, for example), the vesicle membrane needs to be disrupted to allow the loaded agent to come out. For this reason, research on stimuli-responsive BCP vesicles has been increasingly active [38–40]. Basically, such a BCP vesicle can be designed to have its membrane to undergo a chemical reaction such as hydrolysis, or to respond to a signal such as a change in temperature or pH of the surrounding solution, or exposure to a magnetic field or light. It is clear that with a LC-BCP, if the membrane is formed by a LCP, the LC order and the mesophase transition can be exploited to make the resulting vesicle react to an external stimulus in a particular way. For this, rational LC-BCP design is the key.
Nassoy and coworkers made an elegant demonstration [41]. They designed and prepared giant asymmetric BCP vesicles (16 μm in diameter) of which the two leaflets are composed of two diblock copolymers that have the same hydrophilic poly(ethylene glycol) (PEG) but different hydrophobic polymer blocks. As depicted in Figure 8.8, the inner leaflet has polybutadiene that is not responsive to stimuli, while the outer leaflet has a side-on LCP with azobenzene mesogens in the membrane. Upon exposure to UV light inducing the trans–cis isomerization, a LC–isotropic phase transition occurs in the outer leaflet and the associated polymer chain conformational transition, from an extended to a coiled state, results in a contraction of the outer monolayer along the membrane thickness direction (similar to what happens in the shape-memory polymer as discussed above); the excess surface induces a curling instability that leads to the bursting of the vesicle membrane. The authors showed convincing experimental and theoretical results that support the proposed mechanism, for which a photo-induced LC–isotropic phase transition is at the origin.
Figure 8.8 (a) Chemical structures of the two used amphiphilic diblock copolymers. (b) Schematic illustration of a giant asymmetric vesicle formed by the two polymers and the conformational changes within the bilayer membrane upon the trans–cis photoisomerization of azobenzene mesogens. (c) Snapshots showing the bursting of a giant vesicle upon UV light exposure (the first image shows the vesicle before illumination). Source: Mabrouk et al. (2009). Reprinted with permission.

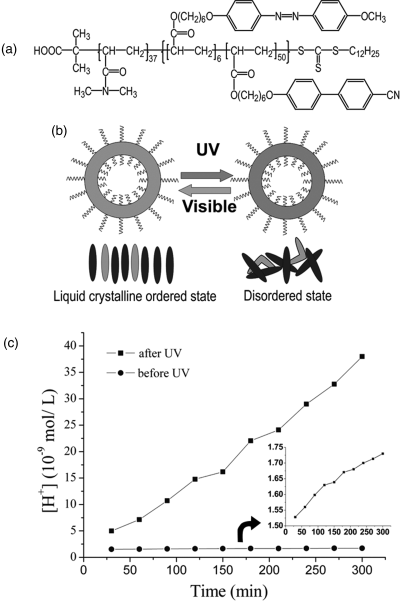
Recently, by designing a new LC-BCP, our group investigated a different approach that makes use of a photo-plasticization effect to soften the vesicle membrane by light instead of bursting it [42]. As shown in Figure 8.9, the idea is to use an amphiphilic LC-BCP whose vesicle membrane-forming LCP bears randomly distributed biphenyl (in majority) and azobenzene mesogens (in minority); and upon absorption of UV light, the trans–cis isomerization of azobenzene could induce a LC–isotropic phase transition exerting a plasticization effect on the membrane. Using a diblock copolymer comprising water-soluble poly(N,N-dimethylacrylamide) (PDMA) and a LC random copolymer containing a small amount of azobenzene mesogens (~12 mol% with respect to biphenyl mesogens), photo-optical measurements on a vesicle solution found evidence that a photo-induced LC order–disorder transition within the vesicle membrane occurred in aqueous solution. Moreover, using a pH-sensitive fluorescent probe, namely, 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS), we found that softening of the vesicle membrane resulting from the photoisomerization led to an increase in the rate of proton diffusion from the interior to outside of the vesicle through the LCP membrane that is similar to plasticization of the vesicle membrane by adding a good solvent in aqueous solution [43].
Figure 8.9 (a) Chemical structures of the amphiphilic LC diblock copolymer. (b) Schematic illustration of a vesicle whose LC membrane can undergo an order–disorder transition induced by the trans–cis photoisomerization of a small number of azobenzene mesogens as a result of the LC co-operative effect. (c) Plot of proton concentration [H+] vs. time for a vesicle solution with a pH gradient at room temperature; the inset is the rescaled result obtained with the solution without UV irradiation [the proton concentration is calculated from the excitation spectra of a pH-sensitive fluorescent probe, namely, 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS)]. Source: Yan et al. (2011). Reprinted with permission.
8.5 Outlook
This chapter highlights the representative examples of the research works in recent years that exploited the interplay of LC order and self-assembled nanostructures in LC-BCPs in such a way that a new function has been generated. Some common features are emerged from these studies. As LCPs, what can be exploited is mainly the great ease of long-range molecular orientation of mesogens. This has been used as an ordering template to induce a macroscopic orientation of cylindrical or strip-like nanodomains in BCPs, and for obtaining a uniform orientation in shape-memory polymers. The transition from an ordered to a disordered state (LC–isotropic phase transformation) is another feature that has been exploited for inducing a reversible polymer chain conformational switch required in shape-memory or actuator materials, and for disrupting the membrane of BCP vesicles. As for nanostructures in BCPs, they are mainly nanocylinders (for macroscopic orientation) and self-assembled network structures with micellar or lamellar nanodomains acting as physical cross-links (for shape-memory, actuator, and thermoplastic elastomers) in the solid state, and are vesicles in solution. In all cases, rational LC-BCP design played a key role in enabling the interaction of LC order and a given nanostructure to result in a target new function. Having identified these common features, it is safe to say that there is still much room for creative LC-BCP design for improving the demonstrated functions or for generating new properties.
LC order can be controlled or configured by an electric or magnetic field. It is conceivable that these external fields could also be exploited for organizing and manipulating the microphase separated structures in LC-BCPs through the stimuli-responsive orientation of mesogens. For instance, the lamellar structure in some BCPs have been investigated as photonic crystals [14] for selective reflection of light. If one domain contains mesogens, a change in their orientation induced by an electric or magnetic field could change the refractive index modulation of the alternating domains and thus result in a wavelength shift for the reflected light. Another possible way to develop novel LC-BCPs to take advantage of the active interaction between LC order and nanostructures would be to design more complex BCP structures. One example is the double LC-BCPs of which the two blocks are LCPs [44]. In the case of the use of a lamellar structure for artificial muscles [27], if the rubbery block is also a side-on or main-chain LCP containing azobenzene moieties (a low Tg is required), the thermally induced contraction and elongation may be more prominent.
1. H. Fischer, S. Poser, M. Arnold, and W. Frank. On the influence of the morphological structure on the liquid crystalline behavior of liquid crystalline side chain block copolymers. Macromolecules 1994, 27, 7133–7138.
2. G. C. L. Wong, J. Commandeur, H. Fischer, and W. H. de Jeu. Orientational wetting in hybrid liquid crystalline block copolymers. Phys. Rev. Lett. 1996, 77, 5221–5224.
3. J. Sanger, W. Gronski, S. Maas, B. Stuhn, and B. Heck. Structural transition in a nematic LC block copolymer induced by the transition to the LC phase. Macromolecules 1997, 30, 6783–6787.
4. G. Mao, J. Wang, S. R. Clingman, and C. K. Ober. Molecular design, synthesis, and characterization of liquid. Macromolecules 1997, 30, 2556–2567.
5. M. Yamada, T. Iguchi, A. Hirao, S. Nakahama, and J. Watanabe. Side-chain liquid crystal block copolymers with well-defined structures prepared by living anionic polymerization I. Thermotropic phase behavior and structures of liquid crystal segment in lamellar type of microphase domain. Polym. J. 1998, 30, 23–30.
6. W. Y. Zheng, R. J. Albalak, and P. T. Hammond. Mesogen orientation within smectic C* side chain liquid crystalline diblock copolymers. Macromolecules 1998, 31, 2686–2689.
7. M. Anthamatten, W. Y. Zheng, and P. T. Hammond. A morphological study of well-defined smectic side-chain LC block copolymers. Macromolecules 1999, 32, 4838–4848.
8. A. Schneider, J.-J. Zanna, M. Yamada, H. Finkelmann, and R. Thomann. Competition between liquid crystalline phase symmetry and microphase morphology in a chiral smectic liquid crystalline–isotropic block copolymer. Macromolecules 2000, 33, 649–651.
9. K. K. Tenneti, X. Chen, C. Y. Li, X. Wan, X. Fan, Q. Zhou, L. Rong, and B. S. Hsia. Competition between liquid crystallinity and block copolymer self-assembly in core–shell rod–coil block copolymers. Soft Matter 2008, 4, 458–461.
10. E. Verploegen, T. Zhang, Y. S. Jung, C. Ross, and P. T. Hammond. Controlling the morphology of side chain liquid crystalline block copolymer thin films through variations in liquid crystalline content. Nano Lett. 2008, 8, 3434–3440.
11. Y. Zhao and J. He. Azobenzene-containing block copolymers: the interplay of light and morphology enables new functions. Soft Matter 2009, 5, 2686–2693.
12. H. F. Yu and T. Ikeda. Photocontrollable liquid–crystalline actuators. Adv. Mater. 2011, 23, 2149–2180.
13. H. Yu, J. Li, T. Ikeda, and T. Iyoda. Macroscopic parallel nanocylinder array fabrication using a simple rubbing technique. Adv. Mater. 2006, 18, 2213–2215.
14. C. Park, J. Yoon, and E. L. Thomas. Enabling nanotechnology with self assembled block copolymer patterns. Polymer 2003, 44, 6725–6760.
15. T. P. Lodge. Block copolymers: past successes and future challenges. Macromol. Chem. Phys. 2003, 204, 265–273.
16. A. Natansohn and P. Rochon. Photoinduced motion in azo-containing polymers. Chem. Rev. 2002, 102, 4139–4175.
17. H. Yu, T. Iyoda, and T. Ikeda. Photoinduced alignment of nanocylinders by supramolecular cooperative motions. J. Am. Chem. Soc. 2006, 128, 11010–11011.
18. Y. Morikawa, T. Kondo, S. Nagano, and T. Seki. Photoinduced 3D ordering and patterning of microphase-separated nanostructure in polystyrene-based block copolymer. Chem. Mater. 2007, 19, 1540–1542.
19. Y. Morikawa, S. Nagano, K. Watanabe, K. Kamata, T. Iyoda, and T. Seki. Optical alignment and patterning of nanoscale microdomains in a block copolymer thin film. Adv. Mater. 2006, 18, 883–886.
20. P. T. Wu, G. Ren, C. Li, R. Mezzenga, and S. A. Jenekhe. Crystalline diblock conjugated copolymers: synthesis, self-assembly, and microphase separation of poly(3-butylthiophene)-b-poly(3-octylthiophene). Macromolecules 2009, 42, 2317–2320.
21. B. W. Boudouris, C. D. Frisbie, and M. A. Hillmyer. Polylactide–polythiophene–polylactide triblock copolymers. Macromolecules 2010, 43, 3566–3569.
22. I. Botiz and S. B. Darling. Self-assembly of poly(3-hexylthiophene)-block-polylactide block copolymer and subsequent incorporation of electron acceptor material. Macromolecules 2009, 42, 8211–8217.
23. D. H. Han, X. Tong, Y. Zhao, and Y. Zhao. Block copolymers comprising p-conjugated and liquid crystalline subunits: induction of macroscopic nanodomain orientation. Angew. Chem. Int. Ed. 2010, 49, 9162–9165.
24. S. V. Ahir, A. R. Tajbakhsh, and E. M. Terentjev. Self-assembled shape-memory fibers of triblock liquid–crystal polymers. Adv. Funct. Mater. 2006, 16, 556–560.
25. A. Lendlein and S. K. Dr. Shape-memory polymers. Angew. Chem. Int. Ed. 2002, 41, 2034–2057.
26. M. Behl and A. Lendlein. Actively moving polymers. Soft Matter. 2007, 3, 58–67.
27. M.-H. Li, P. Keller, J. Yang, and P.-A. Albouy. An artificial muscle with lamellar structure based on a nematic triblock copolymer. Adv. Mater. 2004, 16, 1922–1925.
28. X.-F. Chen, Z. Shen, X.-H. Wan, X.-H. Fan, E.-Q. Chen, Y. Ma, and Q.-F. Zhou. Mesogen-jacketed liquid crystalline polymers. Chem. Soc. Rev. 2010, 39, 3072–3101.
29. P.-G. de Gennes. Un muscle artificiel sem-rapide. C. R. 'Acad. Sci. Ser IIB: Mec Phys Chim Astron 2B 1997, 324, 343–348.
30. M. D. Kempe, N. R. Scruggs, R. Verduzco, J. Lal, and J. A. Kornfield. Self-assembled liquid–crystalline gels designed from the bottom up. Nat. Mater. 2004, 3, 177–182.
31. S. Bai and Y. Zhao. Azobenzene-containing thermoplastic elastomers: Coupling mechanical and optical effects. Macromolecules 2001, 34, 9032–9038.
32. Y. Zhao, S. Bai, D. Dumont, and T. Galstian. Mechanically tunable diffraction gratings recorded on an azobenzene elastomer. Adv. Mater. 2002, 14, 512–514.
33. L. Cui, X. Tong, X. Yan, G. Liu, and Y. Zhao. Photoactive thermoplastic elastomers of azobenzene-containing triblock copolymers prepared through atom transfer radical polymerization. Macromolecules 2004, 37, 7097–7104.
34. J. Yang, D. Lévy, W. Deng, P. Keller, and M.-H. Li. Polymer vesicles formed by amphiphilic diblock copolymers containing a thermotropic liquid crystalline polymer block. Chem. Commun. 2005, 4345–4347.
35. L. Jia, A. Cao, D. Lévy, B. Xu, P.-A. Albouy, X. Xing, M. J. Bowick, and M.-H. Li. Smectic polymer vesicles. Soft Matter 2009, 5, 3446.
36. L. Zhang, A. Eisenberg. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science 1995, 268, 1728–1731.
37. B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher, and D. A. Hammer. Polymersomes: tough vesicles made from diblock copolymers. Science 1999, 284, 1143.
38. A. Blanazs, S. P. Armes, and A. J. Ryan. Self-assembled block copolymer aggregates: from micelles to vesicles and their biological applications. Macromol. Rapid Commun. 2009, 30, 267–277.
39. K. Kita-Tokarczyk, J. Grumelard, T. Haefele, and W. Meier. Block copolymer vesicles-using concepts from polymer chemistry to mimic biomembranes. Polymer 2005, 46, 3540.
40. M. Li and P. Keller. Stimuli-responsive polymer vesicles. Soft Matter 2009, 5, 927–937.
41. E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy, and M.-H. Li. Bursting of sensitive polymersomes induced by curling. PNAS 2009, 106, 7294–7298.
42. B. Yan, X. Tong, P. Ayotte, and Y. Zhao. Light-responsive block copolymer vesicles based on a photo-softening effect. Soft Matter,2011, 7, 10001–10009.
43. J. Wu and A. Eisenberg. Proton diffusion across membranes of vesicles of poly(styrene-b-acrylic acid) diblock copolymers. J. Am. Chem. Soc. 2006, 128, 2880–2884.
44. Y. Zhao, B. Qi, X. Tong and Y. Zhao. Synthesis of Double Side-Chain Liquid Crystalline Block Copolymers Using RAFT Polymerization and The Orientational Cooperative Effect. Macromolecules 2008, 41, 3823–3831.