
8.4. SOx Emissions
Sulfur compounds pose a dual problem. Not only do their combustion products contribute to atmospheric pollution, these products are also so corrosive that they cause severe problems in the operation of gas turbines and industrial power plants. Sulfur pollution and corrosion were recognized as problems long before the nitrogen oxides were known to affect the atmosphere. For a time, the general availability of low-sulfur fuels diminished the general concern with respect to the sulfur. However, the possibility that China is developing its huge coal resources has again raised the specter of massive sulfur oxide emissions. Sulfur may be removed from residual oils by catalytic hydrodesulfurization techniques, but the costs of this process are high and the desulfurized residual oils have a tendency to become waxy at low temperatures. To remove sulfur from coal is an even more imposing problem. It is possible to remove pyrites from coal, but this approach is limited by the size of the pyrite particles.
Unfortunately, pyrite sulfur makes up only half the sulfur content of coal, while the other half is organically bound. Coal gasification is the only means by which this sulfur mode can be removed. Of course, it is always possible to eliminate the deleterious effects of sulfur by removing the major product oxide SO2 by absorption processes. These processes impose large initial capital investments.
The presence of sulfur compounds in the combustion process can affect the nitrogen oxides, as well. Thus, it is important to study sulfur compound oxidation not only to find alternative or new means of controlling the emission of objectionable sulfur oxides, but also to understand their effect on the formation and concentration of other pollutants, especially NOx.
There are some basic differences between the sulfur problem and that posed by the formation of the nitrogen oxides. Nitrogen in any combustion process can be either atmospheric or organically bound. Sulfur can be present in elemental form or organically bound, or it may be present as a species in various inorganic compounds. Once it enters the combustion process, sulfur is reactive with oxidizing species, and in analogy with fuel nitrogen, its conversion to the sulfurous oxides is fast compared with the other energy-releasing reactions.
Although sulfur oxides were recognized as a problem in combustion processes well before the concern regarding photochemical smog and the role of the nitrogen oxides in creating this smog, much less is understood about the mechanisms of sulfur oxidation. Indeed, the amount of recent work on sulfur oxidation has been minimal. The status of the field was reviewed by Levy et al. [42] and Cullis and Mulcahy [43] and much of the material from the following subsections has been drawn from Cullis and Mulcahy's article.
8.4.1. Product Composition and Structure of Sulfur Compounds
When elemental sulfur or a sulfur-bearing compound is present in any combustion system, the predominant product is sulfur dioxide. The concentration of sulfur trioxide found in combustion systems is interesting. Even under lean conditions, the amount of sulfur trioxide formed is only a few percent of that of sulfur dioxide. Generally, however, the sulfur trioxide concentration is higher than its equilibrium value, as would be expected from the relation
![]() (8.92)
(8.92)
These higher-than-equilibrium concentrations may be because the homogeneous reactions that would reduce the SO3 to SO2 and O2 are slow. This point will be discussed later in this section.
It is well known that SO3 has a great affinity for water and that at low temperatures it appears as sulfuric acid H2SO4. Above 500 °C, sulfuric acid dissociates almost completely into sulfur trioxide and water.
Under fuel-rich combustion conditions, in addition to sulfur dioxide, the stable sulfur products are hydrogen sulfide, carbonyl sulfide, and elemental sulfur.
Owing to their reactivity, other oxides of sulfur may appear only as intermediates in various oxidation reactions. These are sulfur monoxide SO, its dimer (SO)2, and disulfur monoxide S2O. Some confusion has attended the identification of these oxides; for example, what is now known to be S2O was once thought to be SO or (SO)2. The most important of these oxides is sulfur monoxide, which is the crucial intermediate in all high-temperature systems. SO is a highly reactive radical whose ground state is a triplet which is electronically analogous to O2. According to Cullis and Mulcahy [43], its lifetime is seldom longer than a few milliseconds. Spectroscopic studies revealed other species in flames, such as CS, a singlet molecule analogous to CO and much more reactive; S2, a triplet analogous to O2 and the main constituent of sulfur vapor above 600 °C; and the radical HS. Johnson et al. [44] calculated the equilibrium concentration of the various sulfur species for the equivalent of 1% SO, in propane−air flames. Their results, as a function of fuel−air ratio, are shown in Figure 8.10. The dominance of SO2 in the product composition for these equilibrium calculations, even under deficient air conditions, should be noted. As reported earlier, practical systems reveal SO3 concentrations that are higher (1–2%) than those depicted in Figure 8.10.

Insight into much that has and will be discussed can be obtained by studying the structures of the various sulfur compounds given in Table 8.3.
8.4.2. Oxidative Mechanisms of Sulfur Fuels
Sulfur fuels characteristically burn with flames that are pale blue, sometimes intensely so. This color comes about from emissions as a result of the very exothermic reaction
![]() (8.93)
(8.93)
and since it is found in all sulfur−fuel flames, this blue color serves to identify SO as an important reaction intermediate in all cases.



Most studies of sulfur−fuel oxidation have been performed using hydrogen sulfide, H2S, as the fuel. Consequently, the following material will concentrate on understanding the H2S oxidation mechanism. Much of what is learned from this mechanism can be applied to understanding the combustion of COS and CS2 as well as elemental and organically bound sulfur.
8.4.2.1. Hydrogen sulfide
Figure 8.11 is a general representation of the explosion limits of H2S/O2 mixtures. This three-limit curve is very similar to that shown for H2/O2 mixtures. However, there is an important difference in the character of the experimental data that determine the H2S/O2 limits. In the H2S/O2 peninsula and in the third limit region, explosion occurs after an induction period of several seconds.
The main reaction scheme for the low-temperature oxidation of H2S, although not known explicitly, would appear to be
![]() (8.94)
(8.94)
![]() (8.95)
(8.95)
![]() (8.96)
(8.96)
![]() (8.97)
(8.97)

The addition of reaction (8.96) to this scheme is necessary because of the identification of S2O in explosion limit studies. More importantly, Merryman and Levy [45] showed in burner studies that S2O occurs upstream from the peak of the SO concentration and that elemental sulfur is present still further upstream in the preignition zone.
The most probable system for the introduction of elemental sulfur is
![]() (8.98)
(8.98)
![]() (8.99)
(8.99)
Given the preignition zone temperatures and overall pressures at which the flame studies were carried out, it does not seem kinetically feasible that the reaction
![]() (8.100)
(8.100)
could account for the presence of S2O. The disproportion of SO would have to give SO2 as well as S2O. Since SO2 is not found in certain experiments where S2O can be identified, disproportion would not be feasible, so reaction (8.96) appears to be the best candidate for explaining the presence of S2O.
Reaction (8.95) is the branching step in the mechanism. It has been suggested that
![]() (8.101)
(8.101)
competes with reaction (8.95), thus determining the second limit. Cullis and Mulcahy [39] suggested the reaction
![]() (8.102)
(8.102)
as the degenerate branching step. The explicit mechanism for forming S2O and its role in flame processes must be considered an uncertainty.
At higher temperatures, the reaction
![]() (8.103)
(8.103)
becomes competitive with reaction (8.96) and introduces O radicals into the system. The presence of O radicals gives another branching reaction, namely,
![]() (8.104)
(8.104)
The branching is held in check by reaction (8.96), which removes SO, and the fast termolecular reaction
![]() (8.105)
(8.105)
which removes both O radicals and SO.
In shock tube studies, SO2 is formed before OH radicals appear. To explain this result, it has been postulated that the reaction
![]() (8.106)
(8.106)
is possible. This reaction and reaction (8.96) give the overall step
![]() (8.107)
(8.107)
Detailed sampling in flames by Sachjan et al. [46] indicates that the H2S is oxidized in a three-step process. During the first stage, most of the H2S is consumed and the products are mainly sulfur monoxide and water. In the second stage, the concentration of SO decreases, the concentration of OH reaches its maximum value, the SO2 reaches its final concentration, and the concentration of the water begins to build as the hydrogen passes through a maximum.
The interpretation given to these results is that during the first stage, the H2S and O2 are consumed mainly by reactions (8.106) and (8.103)
![]() (8.106)
(8.106)
![]() (8.103)
(8.103)
with some degree of chain branching by reaction (8.104)
![]() (8.104)
(8.104)
In the second stage, reaction (8.103) predominates over reaction (8.106) because of the depletion of the H2S; then the OH concentration rises via reaction (8.104) and begins the oxidation of the hydrogen:
![]() (8.108)
(8.108)
Of course, the complete flame mechanism must include
![]() (8.109)
(8.109)
![]() (8.110)
(8.110)
![]() (8.111)
(8.111)
explain the known fact that H2S inhibits the oxidation of hydrogen.
Using laser fluorescence measurements on fuel-rich H2/O2/N2 flames seeded with H2S, Muller et al. [47] determined the concentrations of SH, S2, SO, SO2, and OH in the post-flame gases. From their results and an evaluation of rate constants, they postulated that the flame chemistry of sulfur under rich conditions could be described by the eight fast bimolecular reactions and the two three-body recombination reactions given in Table 8.4.
Reactions 1 and 6 in Table 8.4 were identified as the reactions that control the S2 and SO2 concentrations, respectively, while reactions 2 and 4 control the S concentration with some contribution from reaction 1. Reaction 6 was the major one for the SO flux rate. The three reactions involving H2S were said to play an important role. SH was found to be controlled by reactions 1–5. Because the first eight reactions were fast, it was concluded that they rapidly establish and maintain equilibrium so that the species S, S2, H2S, SH, SO, and SO2 are efficiently interrelated. Thus, relative concentrations, such as those of SO and SO2, can be calculated from thermodynamic considerations at the local gas temperature from the system of reactions

![]() (8.112)
(8.112)
![]() (8.110)
(8.110)
![]() (8.113)
(8.113)
The three-body recombination reactions listed in Table 8.4 are significant in sulfur-containing flames because one provides the homogeneous catalytic recombination of the important H2−O2 chain carriers H and OH via
![]() (8.114)
(8.114)
![]() (8.115)
(8.115)
![]() (8.116)
(8.116)
while the other
![]() (8.117)
(8.117)
is the only major source of SO3 in flames. Under rich conditions, the SO3 concentration is controlled by other reactions with H and SO, that is,
![]() (8.118)
(8.118)
![]() (8.119)
(8.119)
and under lean conditions by
![]() (8.120)
(8.120)
8.4.2.2. Carbonyl sulfide and CS2
Even though there have been appreciably more studies of CS2, COS is known to exist as an intermediate in CS2 flames. Thus, it appears logical to analyze the COS oxidation mechanism first. Both substances show explosion limit curves that indicate that branched-chain mechanisms exist. Most of the reaction studies used flash photolysis; hence, little information exists on what the chain initiating mechanism for thermal conditions would be.
Carbonyl sulfide flames exhibit two zones. In the first zone, carbon monoxide and sulfur dioxide form; and in the second zone, the carbon monoxide is converted into carbon dioxide. Since these flames are hydrogen-free, it is not surprising that the CO conversion in the second zone is rapidly accelerated by adding a small amount of water to the system.
Photolysis initiates the reaction by generating sulfur atoms
![]() (8.121)
(8.121)
The S atom then engenders the chain branching step
![]() (8.122)
(8.122)
![]() (8.123)
(8.123)
![]() (8.103)
(8.103)
At high temperatures, the slow reaction
![]() (8.124)
(8.124)
must also be considered.
Although the initiation step under purely thermally induced conditions such as those imposed by shocks has not been formulated, it is expected to be a reaction that produces O atoms. The high-temperature mechanism would then be reactions (8.103), and (8.122)–(8.124), with termination by the elimination of the O atoms.
For the explosive reaction of CS2, Myerson et al. [48] suggested
![]() (8.125)
(8.125)
as the chain-initiating step. Although the existence of the superoxide, SOO, is not universally accepted, it is difficult to conceive a more logical thermal initiating step, particularly when the reaction can be induced in the 200−300 °C range. The introduction of the CS by reaction (8.125) starts the following chain scheme:
![]() (8.126)
(8.126)
![]() (8.103)
(8.103)
![]() (8.127)
(8.127)
![]() (8.128)
(8.128)
![]() (8.129)
(8.129)
![]() (8.130)
(8.130)
![]() (8.131)
(8.131)
![]() (8.124)
(8.124)
![]()
The high flammability of CS2 in comparison to COS is probably due to the greater availability of S atoms. At low temperatures, branching occurs in both systems via
![]() (8.122)
(8.122)
Even greater branching occurs since in CS2 one has
![]() (8.127)
(8.127)
The comparable reaction for COS is reaction (8.123)
![]() (8.123)
(8.123)
8.4.2.3. Elemental sulfur
Elemental sulfur is found in the flames of all the sulfur-bearing compounds discussed in the previous subsections. Generally, this sulfur appears as atoms or the dimer S2. When pure sulfur is vaporized at low temperatures, the vapor molecules are polymeric and have the formula S8. Vapor-phase studies of pure sulfur oxidation around 100 °C have shown that the oxidation reaction has the characteristics of a chain reaction. In the explosive studies, the reaction must be stimulated by the introduction of O atoms (spark, ozone) for the explosion to proceed.
Levy et al. [42] reported that Semenov suggested the following initiation and branching reactions:
![]() (8.132)
(8.132)
![]() (8.122)
(8.122)
![]() (8.133)
(8.133)
with the products produced by
![]() (8.93)
(8.93)
![]() (8.103)
(8.103)
![]() (8.134)
(8.134)
![]() (8.135)
(8.135)
A unique feature of the oxidation of pure sulfur is that the percentage of SO3 formed is a much larger (about 20%) fraction of the SOx than is generally found in the oxidation of sulfur compounds.
8.4.2.4. Organic sulfur compounds
It is more than likely that when sulfur occurs in a crude oil or in coal (other than the pyrites), it is organically bound in one of the three forms listed in Table 8.3—the thiols, sulfides, or disulfides. The combustion of these compounds is much different from that of other sulfur compounds in that a large portion of the fuel element is a pure hydrocarbon fragment. Thus, in explosion or flame studies, the branched-chain reactions that determine the overall consumption rate or flame speed would follow those chains characteristic of hydrocarbon combustion rather than the CS, SO, and S radical chains which dominate in H2S, CS2, COS, and S8 combustion.
A major product in the combustion of all organic sulfur compounds is sulfur dioxide. Sulfur dioxide has a well-known inhibiting effect on hydrocarbon and hydrogen oxidation and, indeed, is responsible for a self-inhibition in the oxidation of organic sulfur compounds. This inhibition most likely arises from its role in the removal of H atoms by the termolecular reaction
![]() (8.114)
(8.114)
Sulfoxylate ion, a known radical that has been found in H2–O2–SO2 systems, is sufficiently inert to be destroyed without reforming any active chain carrier. In the lean oxidation of the thiols, even at temperatures around 300 °C, all the sulfur is converted to SO2. At lower temperatures and under rich conditions, disulfides form and other products such as aldehydes and methanol are found. The presence of the disulfides suggests a chain-initiating step similar to that of low-temperature hydrocarbon oxidation:
![]() (8.136)
(8.136)
Cullis and Mulcahy reported that this step is followed by
![]() (8.137)
(8.137)
to form the hydrocarbon radical and sulfur dioxide. One must question whether the SO2 in reaction (8.137) is sulfur dioxide. If the O2 strips the sulfur from the RS radical, it is more likely that the SO2 is the sulfur superoxide, which would decompose or react to form SO. The SO is then oxidized to sulfur dioxide as described previously. The organic radical is oxidized, as discussed in Chapter 3. The radicals formed in the subsequent oxidation, of course, attack the original fuel to give the RS radical, and the initiating step is no longer needed.
The SH bond is sufficiently weaker than the CH bonds so that the RS radical would be the dominant species formed. At high temperatures, it is likely that the RS decay leads to the thioaldehyde
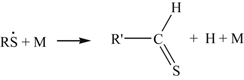 (8.138)
(8.138)The disappearance of the thioaldehyde at these temperatures would closely resemble that of the aldehydes, namely:
 (8.139)
(8.139)![]() (8.140)
(8.140)
Then the CS radical is oxidized, as indicated in the previous discussion on CS2.
The disulfide forms in the thiol oxidation from the recombination of the two RS radicals
![]() (8.141)
(8.141)
The principal products in the oxidation of the sulfides are sulfur dioxide and aldehydes. The low-temperature initiating step is similar to reaction (8.136), except that the hydrogen abstraction is from the carbon atom next to the sulfur atom; that is,
![]() (8.142)
(8.142)
The radical formed in reaction (8.142) then decomposes to form an alkyl radical and a thioaldehyde molecule; that is,
![]() (8.143)
(8.143)
Both products in reaction (8.143) are then oxidized, as discussed.
The oxidation of the disulfides follows a similar route to the sulfide with an initiating step:
![]() (8.144)
(8.144)
followed by radical decomposition
![]() (8.145)
(8.145)
The thiol is then formed by hydrogen abstraction,
![]() (8.146)
(8.146)
and the oxidation proceeds as described previously.
8.4.2.5. Sulfur trioxide and sulfates
As was pointed out earlier, the concentration of sulfur trioxide found in the combustion gases of flames, though small, is greater than would be expected from equilibrium calculations. Indeed, this same phenomenon exists in large combustors such as furnaces, in which there is a sulfur component in the fuel used. The equilibrium represented by Eqn (8.92)
![]() (8.92)
(8.92)
is shifted strongly to the left at high temperatures, so one would expect little SO3 in a real combustion environment. It is readily apparent, then, that the combustion chemistry involved in oxidizing sulfur dioxide to the trioxide is such that equilibrium cannot be obtained.
Truly, the most interesting finding is that the superequilibrium concentrations of SO3 are sensitive to the original oxygen concentration. Under fuel-rich conditions approaching even stoichiometric conditions, practically no SO3 is found. In proceeding from stoichiometric to 1% excess air, a sharp increase in the conversion of SO2 to SO3 is found. Further addition of air causes only a slight increase; however, the effect of the excess nitrogen in reducing the temperature could be a moderating factor in the rate of increase. Figure 8.12, taken from the work of Barrett et al. [49] on hydrocarbon flames, characterizes the results generally found both in flame studies and in furnaces. Such results strongly indicate that the SO2 is converted into SO3 in a termolecular reaction with oxygen atoms:
![]() (8.117)
(8.117)

The superequilibrium results are obtained with sulfur fuels, small concentrations of sulfur fuels added to hydrocarbons, SO2 added to hydrocarbon, and so forth. Further confirmation supporting reaction (8.117) as the conversion route comes from the observation that in carbon monoxide flames the amount of SO3 produced is substantially higher than in all other cases. It is well known that since O atoms cannot attack CO directly, the SO3 concentration is much higher in CO flames than in any other flames. The fact that in all cases the SO3 concentration also increases with pressure supports a termolecular route such as reaction (8.117).
It is well known that the thermal dissociation of SO3 is slow and that the concentration of SO3 is therefore frozen within its stay time in flames and furnaces. The thermal dissociation rates are known, but one can also calculate the superequilibrium concentration of oxygen atoms in flames. If one does so, the SO3 concentration should correspond to the equilibrium concentration given by reaction (8.117), in which the oxygen atom superequilibrium concentration is used. However, the SO3 concentrations are never this high; thus, one must conclude that some SO3 is being reduced by routes other than thermal decomposition. The two most likely routes are by oxygen and hydrogen atom attack on the sulfur trioxide via
![]() (8.120)
(8.120)
![]() (8.119)
(8.119)
Evidence supports this contention as well as the suggestion that reaction (8.120) would be more important than reaction (8.119) in controlling the SO3 concentration with reaction (8.117). Furthermore, one must recognize that reactions (8.117) and (8.120) are effective means of reducing the O radical concentration. Since reaction (8.114) has been shown to be an effective means of reducing H radical concentrations, one can draw the important general conclusion that SO2 and SO3 compounds reduce the extent of superequilibrium concentration of the characteristic chain-carrying radicals which exist in hydrocarbon flames.
In furnaces using residual oils, heterogeneous catalysis is a possible route for the conversion of SO2 to SO3. Sulfur dioxide and molecular oxygen will react catalytically on steel surfaces and vanadium pentoxide (deposited from vanadium compounds in the fuel). Catalytic reactions may also occur at lower temperatures where the equilibrium represented by reaction (8.92) favors the formation of SO3.
If indeed SO2 and SO3 are effective in reducing the superequilibrium concentration of radicals in flames, sulfur compounds must play a role in NO formation from atmospheric nitrogen in flame systems. Since SO2 and SO3 form no matter what type of sulfur compound is added to combustion systems, these species should reduce the oxygen atom concentration and hence should inhibit NO formation. Wendt and Ekmann [50] reported flame data that appear to substantiate this conclusion.
In examining reactions (8.117) and (8.120), one realizes that SO2 plays a role in catalyzing the recombination of oxygen atoms. Indeed, this homogeneous catalytic recombination of oxygen atoms causes the decrease in the superequilibrium concentration of the oxygen atoms. SO2 also plays a role in the recombination of hydrogen radicals through the route
![]() (8.114)
(8.114)
![]() (8.115)
(8.115)
and in the recombination of hydrogen and hydroxyl radicals through the route of reaction (8.114) and
![]() (8.116)
(8.116)
Combining the preceding considerations of SO3 formation with the realization that SO2 and SO are the major sulfur oxide species in flames, one may use the term SOx to specify the sum of SO, SO2, and SO3. Also, considering that in any combustion system sulfur can appear in the parent hydrocarbon fuel in various forms, it becomes evident that the details of the reaction mechanism for SOx formation are virtually impossible to specify. By virtue of the rapidity of the SOx formation process, Bowman [1] argued that the need for a detailed fuel–sulfur oxidation model may be circumvented by postulating approximate models to estimate the gaseous SOx product distribution in the exhaust products. It has been suggested [50] that three principal assumptions would be involved in the proposed model: (1) the fuel−sulfur compounds should be considered minor species so that the major stable species concentrations would be those due to combustion of the hydrocarbon fuel; (2) the bimolecular H2–O2 reactions and reaction (8.113) would be partially equilibrated in the post-flame gases; (3) the SO3 concentration would be calculated from reactions (8.117), (8.119), and (8.120). The SOx pool would then vary as the overall reaction approaches equilibrium by means of the H2–O2 radical recombination reactions, the reactions that determine the equilibrium between SO2 and SO3, and those that affect the catalytic recombination of H, O, and OH.
8.4.2.6. Fuel−sulfur and fuel−nitrogen interactions
The reactions of fuel−sulfur and fuel−nitrogen are closely coupled to the fuel oxidation; moreover, sulfur-containing radicals and nitrogen-containing radicals compete for the available H, O, OH radicals with the hydrocarbons [24]. Because of this close coupling of the sulfur and nitrogen chemistry and the H, O, OH radical pool in flames, interactions between fuel−sulfur and fuel−nitrogen chemistry are to be expected.
The catalytic reduction of the radicals, particularly the O atom, by sulfur compounds will generally reduce the rates of reactions converting atmospheric nitrogen to NO by the thermal mechanism. However, experiments do not permit explicit conclusions [24]. For example, Wendt and Ekmann [50] showed that high concentrations of SO2 and H2S have an inhibiting effect on thermal NO in premixed methene−air flames, while deSoete [51] showed the opposite effect. To resolve this conflict, Wendt et al. [52] studied the influence of fuel−sulfur on fuel−NO in rich flames, whereupon they found both enhancement and inhibition.
A further interaction comes into play when the thermal DeNOx process is used to reduce NOx. When stack gases cool and initial sulfur is present in the fuel, the SO3 that forms reacts with water to form a mist of sulfuric acid, which is detrimental to the physical plant. Furthermore, the ammonia from the thermal DeNOx process reacts with water to form NH4HSO2, a glue-like, highly corrosive compound. These SO3 conditions can be avoided by reducing the SO3 back to SO2. Under stack (post-combustion) temperatures, the principal elementary reactions for SO3 to SO2 conversion are
![]()
![]()
Since the key to this sequence is the HO2 radical, the aqueous hydrogen peroxide process discussed for NO to NO2 conversion in the stack would be an appropriate approach. The SO2 forms no corrosive liquid mist in the stack and could be removed by wet scrubbing of the exhaust.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.