
8.3. Formation and Reduction of Nitrogen Oxides
The previous sections help establish the great importance of the nitrogen oxides in the photochemical smog reaction cycles described. Strong evidence indicated that the major culprit in NOx production was the automobile. But as automobile emissions standards were enforced, attention was directed to power generation plants that use fossil fuels. Given these concerns and those associated with supersonic flight in the stratosphere, great interest remains in predicting—and reducing—nitrogen oxide emissions; this interest has led to the formulation of various mechanisms and analytical models to predict specifically the formation and reduction of nitrogen oxides in combustion systems. This section offers some insight into these mechanisms and models, drawing heavily from the reviews by Bowman [1] and Miller and Bowman [6].
When discussing nitrogen oxide formation from nitrogen in atmospheric air, one refers specifically to the NO formed in combustion systems in which the original fuel contains no nitrogen atoms chemically bonded to other chemical elements such as carbon or hydrogen. Since this NO from atmospheric air forms most extensively at high temperatures, it is generally referred to as thermal NO.
One early controversy with regard to NOx chemistry revolved around what was termed “prompt” NO. Prompt NO was postulated to form in the flame zone by mechanisms other than those thought to hold exclusively for NO formation from atmospheric nitrogen in the high-temperature zone of the flame or post-flame zone. Although the amount of prompt NO formed is small under most practical conditions, the fundamental studies in this problem have helped clarify much about NOx formation and reduction both from atmospheric and fuel-bound nitrogen. The debate focused on the question of whether prompt NO formation resulted from reaction of hydrocarbon radicals and nitrogen in the flame or from nitrogen reactions with large quantities of O atoms generated early in the flame. Furthermore, it was suggested that superequilibrium concentrations of O atoms could, under certain conditions of pressure and stoichiometry, lead to the formation of nitrous oxide, N2O, a subsequent source of NO. These questions are fully addressed later in this section.
The term “prompt” NO derives from the fact that the nitrogen in air can form small quantities of carbon–nitrogen (CN) compounds in the flame zone. In contrast, thermal NO forms in the high-temperature post-flame zone. These CN compounds subsequently react to form NO. The stable compound HCN has been found in the flame zone and is a product in very fuel-rich flames. Chemical models of hydrocarbon reaction processes reveal that early in the reaction, O atom concentrations can reach superequilibrium proportions; and, indeed, if temperatures are high enough, these high concentrations could lead to early formation of NO by the same mechanisms that describe thermal NO formation.
As mentioned, NOx formation from fuel-bound nitrogen is meant to specify the nitrogen oxides formed from fuel compounds that are chemically bonded to other elements. Fuel-bound nitrogen compounds are ammonia, pyridine, and many other amines. The amines can be designated as RNH2, where R is an organic radical or H atom. The NO formed from HCN and the fuel fragments from the nitrogen compounds are sometimes referred to as chemical NO in terminology analogous to that of thermal NO.
Although most early analytical and experimental studies focused on NO formation, more information now exists on NO2 and the conditions under which it is likely to form in combustion systems. Some measurements in practical combustion systems have shown large amounts of NO2, which would be expected under the operating conditions. Controversy has surrounded the question of the extent of NO2 formation in that the NO2 measured in some experiments may actually have formed in the probes used to capture the gas sample. Indeed, some recent high-pressure experiments have revealed the presence of N2O.
8.3.1. Structure of Nitrogen Oxides
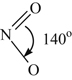
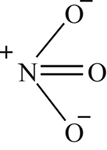
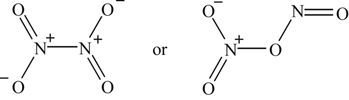
Many investigators have attempted to investigate analytically the formation of NO in fuel−air combustion systems. Given the availability of an enormous amount of computer capacity, they have written all the reactions of the nitrogen oxides they thought possible. Unfortunately, some of these investigators have ignored the fact that some of the reactions could have been eliminated because of steric considerations, as discussed with respect to sulfur oxidation. Since the structure of the various nitrogen oxides can be important, their formulas and structures are given in Table 8.1.

8.3.2. Effect of Flame Structure
As the important effect of temperature on NO formation is discussed in the following sections, it is useful to remember that flame structure can play a significant role in determining the overall NOx emitted. For premixed systems like those obtained on Bunsen and flat flame burners and almost obtained in carbureted spark-ignition engines, the temperature, and hence the mixture ratio, is the prime parameter in determining the quantities of NOx formed. Ideally, as in equilibrium systems, the NO formation should peak at the stoichiometric value and decline on both the fuel-rich and fuel-lean sides, just as the temperature does. Actually, because of kinetic (nonequilibrium) effects, the peak is found on the lean (oxygen-rich) side of stoichiometric.
However, in fuel-injection systems where the fuel is injected into a chamber containing air or an air stream, the fuel droplets or fuel jets burn as diffusion flames, even though the overall mixture ratio may be lean and the final temperature could correspond to this overall mixture ratio. The temperature of these diffusion flames is at the stoichiometric value during part of the burning time, even though the excess species will eventually dilute the products of the flame to reach the true equilibrium final temperature. Thus, in diffusion flames more NOx forms than would be expected from a calculation of an equilibrium temperature based on the overall mixture ratio. The reduction reactions of NO are so slow that in most practical systems the amount of NO formed in diffusion flames is unaffected by the subsequent drop in temperature caused by dilution of the excess species.
8.3.3. Reaction Mechanisms of Oxides of Nitrogen
Nitric oxide is the primary nitrogen oxide emitted from most combustion sources. The role of nitrogen dioxide in photochemical smog has already been discussed. Stringent emission regulations have made it necessary to examine all possible sources of NO. The presence of N2O under certain circumstances could, as mentioned, lead to the formation of NO. In the following subsections the reaction mechanisms of the three nitrogen oxides of concern are examined.
8.3.3.1. Nitric oxide reaction mechanisms
There are three major sources of the NO formed in combustion: (1) oxidation of atmospheric (molecular) nitrogen via the thermal NO mechanisms; (2) prompt NO mechanisms; and (3) oxidation of nitrogen-containing organic compounds in fossil fuels via the fuel-bound NO mechanisms [1]. The extent to which each contributes is an important consideration.
Thermal NO mechanisms: For premixed combustion systems a conservative estimate of the thermal contribution to NO formation can be made by considering the equilibrium system given by reaction (8.48):
![]() (8.48)
(8.48)
As is undoubtedly apparent, the kinetic route of NO formation is not the attack of an oxygen molecule on a nitrogen molecule. Mechanistically, as described in Chapter 3, oxygen atoms form from the H2–O2 radical pool, or possibly from the dissociation of O2, and these oxygen atoms attack nitrogen molecules to start the simple chain shown by reactions (8.49) and (8.50):
![]() (8.49)
(8.49)
![]() (8.50)
(8.50)
where the activation energies are in kJ/mol. Since this chain was first postulated by Zeldovich [7], the thermal mechanism is often referred to as the Zeldovich mechanism. Common practice now is to include the step
![]() (8.51)
(8.51)
in the thermal mechanism, even though the reacting species are both radicals and therefore the concentration terms in the rate expression for this step would be very small. The combination of reactions (8.49)–(8.51) is frequently referred to as the extended Zeldovich mechanism.
If one invokes the steady-state approximation described in Chapter 2 for the N atom concentration and makes the partial equilibrium assumption also described in Chapter 2 for the reaction system,
![]()
one obtains for the rate of formation of NO [8]
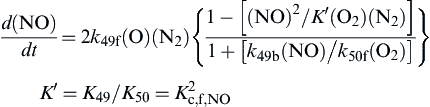 (8.52)
(8.52)
where K is the concentration equilibrium constant for the specified reaction system and K′ the square of the equilibrium constant of formation of NO.
To calculate the thermal NO formation rate from the preceding expression, it is necessary to know the concentrations of O2, N2, O, and OH. But the characteristic time for the forward reaction (8.49) always exceeds the characteristic times for the reaction systems that make up the processes in fuel−oxidizer flame systems; thus, it would appear possible to decouple the thermal NO process from the flame process. Using such an assumption, the NO formation can be calculated from Eqn (8.52) using local equilibrium values of temperature and concentrations of O2, N2, O, and OH.
From examination of Eqn (8.52), one sees that the maximum NO formation rate is given by
![]() (8.53)
(8.53)
which corresponds to the condition that (NO) << (NO)eq. Owing to the assumed equilibrium condition, the concentration of O atoms can be related to the concentration of O2 molecules via

and Eqn (8.53) becomes
![]() (8.54)
(8.54)
The strong dependence of thermal NO formation on the combustion temperature and the lesser dependence on the oxygen concentration are evident from Eqn (8.54). Thus, considering the large activation energy of reaction (8.49), the best practical means of controlling NO is to reduce the combustion gas temperature and, to a lesser extent, the oxygen concentration. For a condition of constant temperature and varying pressure, Eqn (8.53) suggests that the O atom concentration will decrease as the pressure is raised according to Le Chatelier's principle, and the maximum rate will decrease. Indeed, this trend is found in fluidized bed reactors.
To determine the errors that may be introduced by the Zeldovich model, Miller and Bowman [6] calculated the maximum (initial) NO formation rates from the model and compared them with the maximum NO formation rates calculated from a detailed kinetics model for a fuel-rich (ϕ = 1.37) methane−air system. To allow independent variation of temperature, an isothermal system was assumed and the type of prompt NO reactions to be discussed next were omitted. Thus, the observed differences in NO formation rates are due entirely to the nonequilibrium radical concentrations that exist during the combustion process. Their results are shown in Figure 8.1, which indicates a noticeable acceleration of the maximum NO formation rate above that calculated using the Zeldovich model during the initial stages of the reaction owing to nonequilibrium effects, with the departures from the Zeldovich model results decreasing with increasing temperature. As the lower curve in Figure 8.1 indicates, while nonequilibrium effects are evident over a wide range of temperature, the accelerated rates are sufficiently low that little NO is formed by the accelerated nonequilibrium component. Examining the lower curve, as discussed in Chapter 1, one sees that most hydrocarbon−air combustion systems operate in the range of 2100–2600 K.
Prompt NO mechanisms: In dealing with the presentation of prompt NO mechanisms, much can be learned by considering the historical development of the concept of prompt NO. With the development of the Zeldovich mechanism, many investigators followed the concept that in premixed flame systems, NO would form only in the post-flame or burned-gas zone. Thus, it was thought possible to experimentally determine thermal NO formation rates and, from these rates, to find the rate constant of Eqn (8.49) by measurement of the NO concentration profiles in the post-flame zone. Such measurements can be performed readily on flat flame burners. Of course, to make these determinations, it is necessary to know the O atom concentrations. Since hydrocarbon−air flames were always considered, the nitrogen concentration was always in large excess. As discussed in the preceding subsection, the O atom concentration was taken as the equilibrium concentration at the flame temperature and all other reactions were assumed to be very fast compared with the Zeldovich mechanism.

These experimental measurements on flat flame burners revealed that when the NO concentration profiles are extrapolated to the flame-front position, the NO concentration goes not to zero, but to some finite value. Such results were most frequently observed with fuel-rich flames. Fenimore [9] argued that reactions other than the Zeldovich mechanism were playing a role in the flame and that some NO was being formed in the flame region. He called this NO, “prompt” NO. He noted that prompt NO was not found in nonhydrocarbon CO−air and H2−air flames, which were analyzed experimentally in the same manner as the hydrocarbon flames. The reaction scheme he suggested to explain the NO found in the flame zone involved a hydrocarbon species and atmospheric nitrogen. The nitrogen compound was formed via the following mechanism:
![]() (8.55)
(8.55)
![]() (8.56)
(8.56)
The N atoms could form NO, in part at least, by reactions (8.50) and (8.51), and the CN could yield NO by oxygen or oxygen atom attack. It is well known that CH exists in flames and indeed, as stated in Chapter 4, is the molecule that gives the deep violet color to a Bunsen flame.
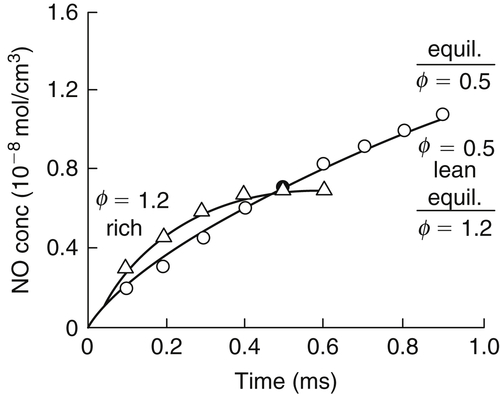
To verify whether reactions other than the Zeldovich mechanism were effective in NO formation, various investigators undertook the study of NO formation kinetics by use of shock tubes. The primary work in this area was that of Bowman and Seery [10], who studied the CH4–O2–N2 system. Complex kinetic calculations of the CH4–O2–N2 reacting system based on early kinetic rate data at a fixed high temperature and pressure similar to those obtained in a shock tube [11] for T = 2477 K and P = 10 atm are shown in Figure 8.2. Even though more recent kinetic rate data would modify the product−time distribution somewhat, the general trends of the product distribution are important and are relatively unaffected by some changes in rates. These results are worth considering in their own right, for they show explicitly much that has been implied. Examination of Figure 8.2 shows that at about 5 × 10−5 s, all the energy–release reactions will have equilibrated before any significant amounts of NO have formed; and, indeed, even at 10−2 s the NO has not reached its equilibrium concentration for T = 2477 K. These results show that for such homogeneous or near-homogeneous reacting systems, it would be possible to quench the NO reactions, obtain the chemical heat release, and prevent NO formation. This procedure has been put into practice in certain combustion schemes.

Equally important is the fact that Figure 8.2 reveals large overshoots within the reaction zone. If these occur within the reaction zone, the O atom concentration could be orders of magnitude greater than its equilibrium value, in which case this condition could lead to the prompt NO found in flames. The mechanism analyzed to obtain the results depicted in Figure 8.2 was essentially that given in Chapter 3, Section 3.7.2, with the Zeldovich reactions. Thus, it was thought possible that the Zeldovich mechanism could account for the prompt NO.
The early experiments of Bowman and Seery appeared to confirm this conclusion. Some of their results are shown in Figure 8.3. In this figure the experimental points compared well with the analytical calculations based on the Zeldovich mechanisms alone. The same computational program as that of Martenay [11] was used. Figure 8.3 also depicts another result frequently observed: fuel-rich systems approach NO equilibrium much faster than do fuel-lean systems [12].
Although Bowman and Seery’s results would seem at first to refute the suggestion by Fenimore that prompt NO forms by reactions other than the Zeldovich mechanism, one must remember that flames and shock tube–initiated reacting systems are distinctively different processes. In a flame there is a temperature profile that begins at the ambient temperature and proceeds to the flame temperature. Thus, although flame temperatures may be simulated in shock tubes, the reactions in flames are initiated at much lower temperatures than those in shock tubes. As stressed many times before, the temperature history frequently determines the kinetic route and the products. Therefore, shock tube results do not prove that the Zeldovich mechanism alone determines prompt NO formation. The prompt NO could arise from other reactions in flames, as suggested by Fenimore.

Bachmeier et al. [13] appeared to confirm Fenimore’s initial postulates and to shed greater light on the flame NO problem. These investigators measured the prompt NO formed as a function of equivalence ratio for many hydrocarbon compounds. Their results are shown in Figure 8.4. What is significant about these results is that the maximum prompt NO is reached on the fuel-rich side of stoichiometric, remains at a high level through a fuel-rich region, and then drops off sharply at an equivalence ratio of about 1.4.
Bachmeier et al. also measured the HCN concentrations through propane−air flames. These results, which are shown in Figure 8.5, show that HCN concentrations rise sharply somewhere in the flame, reach a maximum, and then decrease sharply. However, for an equivalence ratio of 1.5, a fuel-rich condition for which little prompt NO is found, the HCN continues to rise and is not depleted. The explanation offered for this trend is that HCN forms in all the rich hydrocarbon flames; however, below an equivalence ratio of 1.4, O radicals are present in sufficient abundance to deplete HCN and form the NO. Since the sampling and analysis techniques used by Bachmeier et al. [13] did not permit the identification of the cyanogen radical CN, the HCN concentrations found most likely represent the sum of CN and HCN as they exist in the flame. The CN and HCN in the flame are related through the rapid equilibrium reactions [14].

![]() (8.57)
(8.57)
![]() (8.58)
(8.58)
From other more recent studies of NO formation in the combustion of lean and slightly rich methane−oxygen−nitrogen mixtures as well as lean and very rich hydrocarbon−oxygen−nitrogen mixtures, it must be concluded that some of the prompt NO is the result of the overshoot of O and OH radicals above their equilibrium values, as the Bowman and Seery results suggested. But even though O radical overshoot is found on the fuel-rich side of stoichiometric, this overshoot cannot explain the prompt NO formation in fuel-rich systems. It would appear that both the Zeldovich and Fenimore mechanisms are feasible.
Some interesting experiments by Eberius and Just [16] seem to clarify what is happening in the flame zone with regard to NO formation. Eberius and Just's experiments were performed on a flat flame burner with propane as the fuel. Measurements were made of the prompt NO at various fuel−oxygen equivalence ratios whose flame temperatures were controlled by dilution with nitrogen. Thus, a range of temperatures could be obtained for a given propane−oxygen equivalence ratio. The results obtained are shown in Figure 8.6. The highest temperature point for each equivalence ratio corresponds to zero dilution.
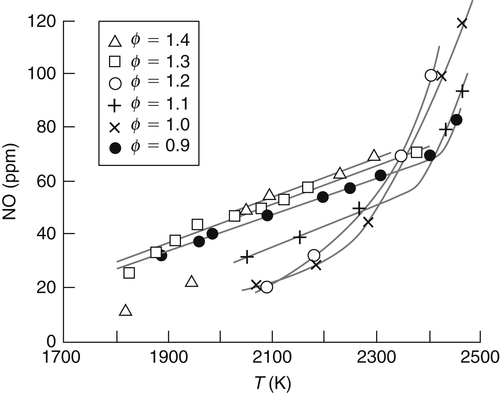
The shapes of the plots in Figure 8.6 are revealing. At both the low- and high-temperature ends, all the plots seem nearly parallel. The slopes at the low-temperature end are much less than the slopes at the high-temperature end, thereby indicating two mechanisms for the formation of prompt NO. The two mechanisms are not related solely to the fuel-rich and fuel-lean stoichiometry, as many investigators thought, but also to the flame temperature. These results suggest two routes: a high-temperature, high-activation route and a lower-temperature, low-activation route.
The systematic appearance of these data led Eberius and Just to estimate the activation energy for the two regions. Without correcting for diffusion, they obtained an activation energy of the order of 270 kJ/mol for the high-temperature zone. This value is remarkably close to the 315 kJ/mol activation energy required for the initiating step in the Zeldovich mechanism. Furthermore, diffusion corrections would raise the experimental value somewhat. The low-temperature region has an activation energy of the order of 50–60 kJ/mol. As will be shown later, radical attack on the cyano species is faster than oxygen radical attack on hydrogen. The activation energy of O + H2 is about 33 kJ/mol; therefore, the HCN reaction should be less. Again, diffusion corrections for the oxygen atom concentration could lower the apparent activities of activation energies of 50–60 kJ/mol to below 34 kJ/mol. This crude estimate of the activation energy from Eberius and Just’s low-temperature region, together with the formation of HCN found by the same group [13] in their other flame studies with propane (Figure 8.5), appears to indicate that the Fenimore mechanism [9] would hold in the lower-temperature region.
The kinetic details for prompt NO formation must begin with the possible reactions between N2 and hydrocarbon fragments, as Fenimore [9] originally suggested. Hayhurst and Vance [17] suggested that two other likely candidate reactions may be added to those posited by Fenimore. The four candidate reactions would then be
![]() (8.59)
(8.59)
![]() (8.60)
(8.60)
![]() (8.61)
(8.61)
![]() (8.62)
(8.62)
As discussed in the introduction to Chapter 4, the existence of C2 and CH in hydrocarbon−air flames is well established. Methylene (CH2) arises in most combustion systems by OH and H attack on the methyl radical (CH3). Similar attack on CH2 creates CH.
Hayhurst and Vance [17] established that the amount of prompt NO in moderately fuel-rich systems is proportional to the number of carbon atoms present per unit volume and is independent of the original parent hydrocarbon identity. This result indicates that reactions (8.59) and (8.60) are not primary contributors because it is unlikely that C2 or C2H could derive from CH4 with an efficiency one-half that from C2H2 or one-third that from C3H6 or C3H8. In their complete model Miller and Bowman [6] introduced the reactions
![]() (8.63)
(8.63)
and
![]() (8.64)
(8.64)
They concluded from estimated rates that reaction (8.63) is an insignificant contributor to prompt NO. However, they also pointed out that the reverse reaction (8.64) is fast at room temperature and under shock tube conditions. Hence, this step is a minor, but nonnegligible contributor to prompt NO, and because of the large endothermicity of reaction (8.64), its importance with respect to reaction (8.61) increases with increasing temperature.
The major products of reactions (8.61) and (8.62) are HCN and NH. In the experimental work of Bachmeier et al. [13], HCN and CN were indistinguishable because of the equilibrium reactions (8.57) and (8.58) mentioned and the analysis methods applied. In consideration of the work of Ref. [1], the major kinetic route to prompt NO would then appear to be
![]() (8.65)
(8.65)
![]() (8.66)
(8.66)
![]() (8.67)
(8.67)
![]() (8.68)
(8.68)
![]() (8.69)
(8.69)
Following the conclusions of Bowman [1], then, from the definition of prompt NO, these sources of prompt NO in hydrocarbon fuel combustion can be identified: (1) nonequilibrium O and OH concentrations in the reaction zone and burned gas, which accelerate the rate of the thermal NO mechanism; (2) a reaction sequence, shown in Figure 8.7, that is initiated by reactions of hydrocarbon radicals, present in and near the reaction zone, with molecular nitrogen (the Fenimore prompt−NO mechanism); and (3) reaction of O atoms with N2 to form N2O via the three-body recombination reaction
![]()
and the subsequent reaction of the N2O to form NO via
![]()
The relative importance of these three mechanisms in NO formation and the total amount of prompt NO formed depend on conditions in the combustor. Acceleration of NO formation by nonequilibrium radical concentrations appears to be more important in non-premixed flames, in stirred reactors for lean conditions, and in low-pressure premixed flames, accounting for up to 80% of the total NO formation. Prompt NO formation by the hydrocarbon radical−molecular nitrogen mechanism is dominant in fuel-rich premixed hydrocarbon combustion and in hydrocarbon diffusion flames, accounting for greater than 50% of the total NO formation. Nitric oxide formation by the N2O mechanism increases in importance as the fuel−air ratio decreases, as the burned gas temperature decreases, or as pressure increases. The N2O mechanism is most important under conditions where the total NO formation rate is relatively low [1].

Bozzelli and Dean [18] proposed that NO can also form through the formation of NNH radicals and their subsequent oxidation via the reaction sequence
![]()
![]()
Experimental evidence for this mechanism has been found in low-pressure H2/air flames, fuel-rich H2/O2/N2 and CH4/O2/N2 flames at atmospheric pressure, and lean H2–air mixtures in stirred reactors. Modeling of these systems by Konnov et al. [19] supports the importance of this mechanism. Through modeling and experimental studies of jet stirred reactors fuels with various hydrocarbons, Ruter et al. [20] found that prompt and NNH pathways dominate the formation of NO in the flame, whereas Zeldovich and nitrous oxide are primary contributors to the NO formation in the post flame. While some uncertainty exists in the NNH rate constants, the NNH mechanism appears to play a role in NO formation.
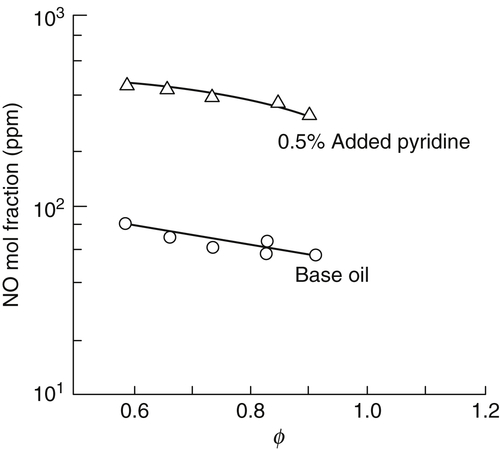
Fuel-bound nitrogen NO mechanisms: In several experiments, it was shown that NO emissions from combustion devices that operate with nitrogen-containing compounds in the fuel are high; in other words, fuel-bound nitrogen is an important source of NO. The initial experiments of Martin and Berkau [21] commanded the greatest interest. These investigators added 0.5% pyridine to base oil and found almost an order of magnitude increase over the NO formed from base oil alone. Their results are shown in Figure 8.8.
During the combustion of fuels containing bound nitrogen compounds, the nitrogen compounds most likely undergo some thermal decomposition prior to entering the combustion zone. Hence, the precursors to NO formation will, in general, be low-molecular-weight, nitrogen-containing compounds or radicals (NH3, NH2, NH, HCN, CN, etc.). All indications are that the oxidation of fuel-bound nitrogen compounds to NO is rapid and occurs on a time scale comparable to the energy–release reactions in the combustion systems. This conclusion arises from the fact that the NH and CN oxidation reactions discussed in the previous section are faster [22,23] than the important chain branching reaction
![]() (8.70)
(8.70)
Thus, the reaction system cannot be quenched to prevent NO formation from fuel-bound nitrogen, as is the case with atmospheric nitrogen. In fact, in the vicinity of the combustion zone, observed NO concentrations significantly exceed calculated equilibrium values. In the post-combustion zone, the NO concentration decreases relatively slowly for fuel-lean mixtures and more rapidly for fuel-rich mixtures. Recall Bowman and Seery's results (Figure 8.3) showing that fuel-rich systems approach equilibrium faster. When fuel−nitrogen compounds are present, high NO yields are obtained for lean and stoichiometric mixtures and relatively lower yields are found for fuel-rich mixtures. The NO yields appear to depend only slightly on temperature, thus indicating a low activation energy step. This result should be compared to the strong temperature dependence of NO formation from atmospheric nitrogen.
The high yields on the lean side of stoichiometric pose a dilemma. It is desirable to operate lean to reduce hydrocarbon and carbon monoxide emissions; but with fuel containing bound nitrogen, high NO yields would be obtained. The reason for the superequilibrium yields is that the reactions leading to the reduction of NO to its equilibrium concentration, namely
![]() (8.71)
(8.71)
![]() (8.72)
(8.72)
![]() (8.73)
(8.73)
are slow. NO can be reduced under certain conditions by CH and NH radicals, which can be present in relatively large concentrations in fuel-rich systems. These reduction steps and their application will be discussed later.
The extent of conversion of fuel nitrogen to NO is nearly independent of the parent fuel molecule, but is strongly dependent on the local combustion environment and on the initial fuel nitrogen in the reactant. Unlike sulfur in the fuel molecule, nitrogen is much more tightly bound in the molecule and, for the most part, in an aromatic ring [24]. Regardless, all fuel−nitrogen compounds exhibit solely carbon−nitrogen or nitrogen−hydrogen bonding. Thus, it is not surprising that in the oxidation of fuel−nitrogen compounds, the major intermediates are HCN and CN and amine radicals stemming from an ammonia structure: that is, NH2, NH, and N.
In a large radical pool, there exists an equilibrium
![]() (8.74)
(8.74)
which essentially establishes an equilibrium between all NH compounds; that is,
![]() (8.75)
(8.75)
![]() (8.76)
(8.76)
Thus, there is great similarity between the prompt NO reactions discussed and the fuel−nitrogen reactions.
In the combustion of fuel−nitrogen compounds, the equilibrium of
![]() (8.57)
(8.57)
will certainly exist. Thus, the conversion of all relevant intermediates to NO has essentially been discussed, and only the NH2 reactions remain to be considered. These reactions follow the sequence
![]() (8.77)
(8.77)
![]() (8.78)
(8.78)
![]() (8.79)
(8.79)
The general scheme of the fuel−nitrogen reactions is also represented in Figure 8.7.
In fuel-rich systems, there is evidence [8,25,26] that the fuel−nitrogen intermediate reacts not only with oxidizing species in the manner represented, but also competitively with NO (or another nitrogen intermediate) to form N2. This second step, of course, is the reason that NO yields are lower in fuel-rich systems. The fraction of fuel nitrogen converted to NO in fuel-rich systems can be as much as an order of magnitude less than that of lean or near-stoichiometric systems. One should realize, however, that even in fuel-rich systems, the exhaust NO concentration is substantially greater than its equilibrium value at the combustion temperature.
Haynes et al. [14] showed that when small amounts of pyridine are added to a premixed, rich (ϕ = 1.68; T = 2030 K) ethylene−air flame, the amount of NO increases with little decay of NO in the post-flame gases. However, when larger amounts of pyridine are added, significant decay of NO is observed after the reaction zone. When increasingly higher amounts of pyridine are added, high concentrations of NO leave the reaction zone, but this concentration drops appreciably in the post-flame gases to a value characteristic of the flame, but well above the calculated equilibrium value. Actual experimental results are shown in Figure 8.9.

In fuel-rich systems, the conversion reactions of the fuel−nitrogen intermediates are subject to doubt, mainly because the normal oxidizing species O2, O, and OH are present only in small concentrations, particularly near the end of the reaction zone. Haynes et al. [14] offered the interesting suggestion that the CN can be oxidized by CO2 since the reaction
![]() (8.80)
(8.80)
is 85 kJ/mol exothermic and estimated to be reasonably fast.
8.3.3.2. Nitrogen dioxide reaction mechanisms
Significant concentrations of NO2 have been reported in the exhaust of gas turbines and in the products of range-top burners [24]. These results are surprising because chemical equilibrium considerations reveal that the NO2/NO ratio should be negligibly small for typical flame temperatures. Furthermore, when kinetic models are modified to include NO2 formation and reduction, they show that the conversion of NO to NO2 can be neglected in practical devices.
However, in the case of sampling from gas turbines, NO2 can vary from 15% to 50% of the total NOx, depending on the NO level [24,27,28]. In the case of range-type burners, the NOx has been reported as high as 15−20 times the NO levels in parts of the flame surrounding the burner top [29,30].
Merryman and Levy [31] examined both NO and NO2 formation in a flat flame burner operated near stoichiometric. In the low-temperature regime of visible flames, they found large concentrations of HO2 that can react with the NO formed in the high-temperature regime and diffuse back to the lower-temperature zone. Their measurements showed that NO2 is produced in the visible regime of all air flames (with and without fuel-bound nitrogen) and that NO is observed only in the visible region when fuel-bound nitrogen is present. Furthermore, these investigators found that NO2 is consumed rapidly in the near–post-flame zone, whereupon the NO concentration rises correspondingly. They postulated the following scheme to represent their findings:
![]() (8.81)
(8.81)
![]() (8.82)
(8.82)
![]() (8.83)
(8.83)
In light of Figure 8.7, whether reaction (8.81) should be the representative reaction for NO formation is irrelevant.
The significant step is represented by reaction (8.82). One should recall that there can be appreciable amounts of HO2 in the early parts of a flame. The appearance of the NO2 is supported further by the fact that reaction (8.82) is two orders of magnitude faster than reaction (8.83). The importance of the hydroperoxy radical attack on NO appeared to be verified by the addition of NO to the cold-fuel mixtures in some experiments. In these tests, the NO disappeared before the visible region was reached in oxygen-rich and stoichiometric flames: that is, flames that would produce HO2. The NO2 persists because, as mentioned previously, its reduction to N2 and O2 is slow. The role of HO2 would not normally be observed in shock tube experiments owing to the high temperatures at which they usually operate.
The Merryman−Levy sequence could explain the experimental results that show high NO2/NO ratios. For the experiments in which these high ratios were found, it is possible that reaction (8.83) is quenched, in which case the NO2 is not reduced. Cernansky and Sawyer [32], in experiments with turbulent diffusion flames, also concluded that the high levels of NO2 found were due to the reactions of NO with HO2 and O atoms.
The experimental efforts reporting high NO2 levels have come into question because of the possibility that much of the NO2 actually forms in sampling tubes [33,34]. Optical techniques are now being applied; but unfortunately, the low concentrations of NO2 that exist make resolution of the controversy difficult.
8.3.3.3. Nitrous oxide formation mechanisms
Quoting directly from Bowman [1], the principal gas-phase reactions forming N2O in fossil fuel combustion are
![]()
![]()
In natural gas combustion an increasingly important contribution from
![]()
occurs in fuel-lean mixtures and at low temperature and elevated pressures. The primary N2O removal steps are
![]()
and
![]()
Calculated lifetimes of N2O in combustion products indicate that for temperatures above 1500 K, the lifetime of N2O typically is less than 10 ms, suggesting that except for low-temperature combustion, as found in fluidized bed combustors and in some post-combustion NO removal systems, N2O emissions should not be significant, a conclusion that is in agreement with the most recent measurements of N2O emissions from combustion devices.
8.3.4. Reduction of NOX
Because of the stringent emissions standards imposed on both mobile and stationary power sources, methods for reducing NOx must be found; moreover, such methods should not impair the efficiency of the device. The simplest method of reducing NOx, particularly from gas turbines, is by adding water to the combustor. Water vapor can reduce the O radical concentration by the following scavenging reaction:
![]() (8.84)
(8.84)
Fortunately, OH radicals do not attack N2 efficiently. However, it is more likely that the effect of water on NOx emissions is through the attendant reduction in combustion temperature. NOx formation from atmospheric nitrogen arises primarily from the temperature-sensitive Zeldovich mechanism.
The problem of NOx reduction is more difficult in heterogeneous systems such as those which arise from direct liquid fuel injection and are known to burn as diffusion flames. One possible means is to decrease the average droplet size formed from injection. Kesten [35] and Bracco [36] showed that the amount of NO formed from droplet diffusion flames can be related to the droplet size: One large droplet will give more NO than can be obtained from a group of smaller droplets whose mass is equal to that of the larger droplet. Any means of decreasing the heterogeneity of a flame system will decrease the NOx. Another possible practical scheme is to emulsify the fuel with a higher vapor pressure, nonsoluble component such as water. It has been shown [37] that droplets from such emulsified fuels explode after combustion has been initiated. These microexplosions occur when the superheated water within the fuel droplet vaporizes, hence appreciably decreasing the heterogeneity of the system. A further benefit is obtained not only because the water is available for dilution, but also because the water is present in the immediate vicinity of the diffusion flame.
If it is impossible to reduce the amount of NOx in the combustion section of a device, the NOx must be removed somewhere in the exhaust. Myerson [38] showed that it is possible to reduce NOx by adding small concentrations of fuel and oxygen. The addition of about 0.1% hydrocarbon (isobutane) and 0.4% O2 to an NOx-containing system at 1260 K reduced the NOx concentration by a factor of two in about 125 ms. Myerson [38] found that the ratio of O2/HC was most important. When the concentrations of O2 and the hydrocarbon are large, an HCN-formation problem could arise. This procedure is feasible only for slightly fuel-lean or fuel-rich systems. The oxygen is the creator and the destroyer of other species involved in the NO reduction. This fact, in turn, means that the initial addition of O2 to the hydrocarbon–NO mixture promotes the production of the strongly reducing species CH and CH2 and similar substituted free radicals that otherwise must be produced by slower pyrolysis reactions.
Continued addition of O2 beyond one-half the stoichiometric value with the hydrocarbons present encourages a net destruction of the hydrocarbon radicals. For the temperature range 1200–1300 K, production of the hydrocarbon radicals via hydrogen abstraction by O2 is rapid, even assuming an activation energy of 520 kJ/mol, and more than adequate to provide sufficient radicals for NO reduction in the stay time range of 125 ms.
Myerson postulated that the following reactions are involved:
![]() (8.85)
(8.85)
![]() (8.86)
(8.86)
The exothermicity of reaction (8.85) is sufficient to fragment the formyl radical and could be written as
![]() (8.87)
(8.87)
In the absence of O2, the N radicals in these fuel-rich systems can react rapidly with NO via
![]() (8.88)
(8.88)
Another technique currently in practice is known as the thermal DeNOx process [39], which uses ammonia as the NOx reduction agent. The ammonia is injected into the exhaust gases of stationary power plants burning fossil fuels. The process is effective in a narrow temperature range, about T ∼ 1250 K. Below about 1100 K, the reaction takes place too slowly to be of value, and above 1400 K more NO is formed. Miller et al. [40] discovered that if H2 is added to the system, the center of the temperature window moves to a lower value without changing the width of the window. They also found that slightly lean combustion products appear to be required for the reduction reaction to be effective; that is, the process is implemented under excess oxygen conditions. Increasing the NH3 concentration to a comparable O2 concentration inhibits the process under certain conditions, and the presence of water slightly inhibits the NO reduction because the optimum temperature is increased slightly [6].
Explaining these effects has been one of the successes of kinetic modeling [6]. The ammonia in the process is considered to form the amine radical NH2, which reacts with the NO to form an intermediate that decays into products:
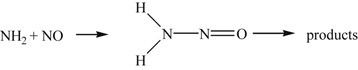 (8.89)
(8.89)There are various possibilities as to the fate of the intermediate. It could form HNNO + H. However, this route is unlikely because no H atoms are found at low temperatures, nor is there any N2O, which would inevitably have to form. In addition, this decay step is endothermic. Another possibility is the formation of N2O and H2, which is exothermic. But there is a large energy barrier involved in forming H2. Also, of course, no N2O is found. The formation of H2N N and O is endothermic and is not conceivable.
N and O is endothermic and is not conceivable.
The possibility exists of a migration of an H atom. Because the migration step
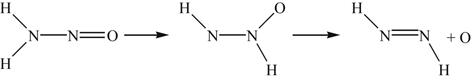 (8.90)
(8.90)is also endothermic, it is ruled out. What appears to be most feasible is the migration of H to the O atom in the following step:
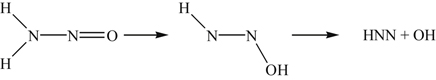 (8.91)
(8.91)The product HNN provides a plausible route for the overall NO reduction mechanism, which permits the determination of the temperature window. Miller and Bowman [6] proposed the competitive channels shown in Table 8.2 as the explanation.
The thermal DeNOx system removes NO in practical systems because the NH2 + NO initiates a significant chain branching system, thereby allowing the overall reaction sequence to be self-sustaining. Following the general scheme in Table 8.2, the conversion of NH3 to NH2 occurs principally by reaction with OH:
![]()

But, in the absence of water vapor, it may occur by reaction with O atoms:
![]()
The required chain branching to regenerate OH and O and hence to continue the conversion is accomplished [6] by the reaction sequence for Channel 1:
![]()
![]()
![]()
The H atom produced in the last step reacts with O2 in the familiar chain branching step
![]()
giving an overall chain branching radical pool. In the presence of the water inherent in the product composition of most combustion systems, the system is further augmented by the reaction of O atoms with water via
![]()
and OH becomes the dominant species converting NH3 to NH2.
In their model, Miller and Bowman [6] consider the important chain termination steps to be
![]()
![]()
and
![]()
They conclude that at the low-temperature end of the effective temperature window, the NO reduction effectiveness is limited principally by the rates of the chain termination reactions that compete with the preceding branching sequence. In addition, below about 1100 K, hydrogen abstraction by OH is so slow that little NH2 forms and H + O2 + M → HO2 + M becomes a competitive step for H + O2 → OH + O. In the temperature range 1100–1400 K, the mix of branching and termination is proper for the conversion of NH3 to NH2 at a sufficiently rapid and sustaining rate. However, above about 1400 K, Channel 2 of Table 8.2 becomes dominant. At such temperatures, there is more chain branching, which leads to a higher concentration of OH. Thus, the Channel 2 reaction
![]()
is favored over the Channel 1 reaction
![]()
These two reactions are competitive around 1250 K. Note that in Channel 1, two NO molecules react to form one N2 and one NO so that an overall reduction in NO is obtained. The role of added H2 manifests itself by increasing the amount of chain branching at the lower temperatures and increasing the overall concentration of OH. Thus, the window shifts to lower temperatures.
Other post-combustion NOx removal techniques include the injection of urea ([NH2]2CO) and cyanuric acid ([HOCN]3). The latter is termed the RapreNOx process [41]. When heated, cyanuric acid sublimes and decomposes to form isocyanic acid HNCO. Similarly, urea reacts to form NH3 and HNCO. Thus, its NOx reduction path follows that of the thermal DeNOx route as well as that of the RapreNOx route to be discussed.
The major route in the RapreNOx process is the radical attack on the isocyanuric acid by H and OH via
![]()
![]()
The first of these two steps forms the amine radical NH2 and it acts as in the thermal DeNOx process. The importance of the CO is that its oxidation produces H atoms from the well-known step
![]()
The importance of the second step is that it provides the primary NO removal step [6]
![]()
for the process. The N2O decays, as discussed earlier, to form N2. The advantage of the RapreNOx process may be its ability to remove NOx at much lower temperatures than the thermal DeNOx process. There is some indication that this lower-temperature effect may be due to decomposition of cyanuric acid on surfaces.
Considering that wet scrubbers are in place in many facilities and more are planned for the future, another efficient means for NOx removal could be considered. These scrubbing methods are limited by the relatively inert nature of NO. It has been proposed that this difficulty can be overcome by the conversion of NO to the much more active NO2 through reaction (8.82), as discussed earlier:
![]()
The question arises as to how to produce HO2 radicals. For post-combustion conversion, the obvious candidate would be an aqueous solution of hydrogen peroxide H2O2. Although hydrogen peroxide readily converts to water and oxygen through a heterogeneous decomposition, its homogeneous decomposition route follows the simple steps:
![]()
![]()
to form the necessary hydroperoxy radical HO2. However, one must realize that this simple reaction sequence is so slow that it is ineffective below 600 K. At high temperatures, particularly at atmospheric pressure, HO2 dissociates. Moreover, it would appear that at temperatures above 1100 K, the general radical pool is large enough that recombination reactions become too competitive. Thus, this aqueous process has an effective temperature window between 600 and 1100 K [6].
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.