
9.5. Practical Carbonaceous Fuels (C. R. Shaddix)
Solid carbonaceous fuels such as coal, biomass, and petroleum coke are widely used throughout the world to provide heat and to generate electrical power through combustion processes. These fuels are generally classified according to their heating value (energy content), volatile fuel content, fixed carbon content, moisture level, and ash or mineral content. The volatile fuel content is the fraction of the original fuel mass that evolves as gases when the fuel is heated to high temperatures, whereas the fixed carbon is the mass of carbon that remains after the volatiles have escaped. The ash content refers to the portion of the solid that remains when the fixed carbon has been fully oxidized, leaving behind oxidized mineral compounds. For some coal and biomass sources, the ash content can be very large and pose a severe hindrance to the effective combustion of the fuel. Furthermore, the formation of ash deposits, particularly those that melt (referred to as “slagging” deposits) or otherwise are difficult to remove, restricts the effective heat transfer from furnace gases to boiler tubes and plays an important role in boiler design and operation. Excessive moisture in the fuel reduces the fuel heating value and flame stability. Conversely, insufficient fuel moisture can lead to spontaneous ignition problems when storing and handling some reactive fuels.
9.5.1. Devolatilization
The devolatilization process (referring to the release of gaseous fuel components as the solid fuel is heated) is a key characteristic of the combustion of solid fuels. The volatile gases burn much more rapidly than the remaining char particles and therefore are important for flame ignition and stability and play an important role in the formation of oxides of nitrogen (NOx), a regulated class of pollutants. Moreover, the devolatilization process determines how much char remains to be burned as well as the physical characteristics of the resulting char, with subsequent impacts on the char combustion properties. Different coal types vary significantly in their volatile content, ranging from a maximum of approximately 50% (by mass) for low- and mid-rank coals to just a few percent for anthracitic coals (which are graphite-like in character). Biomass always has a large volatile content, generally around 80%, as determined using the standard ASTM test method.
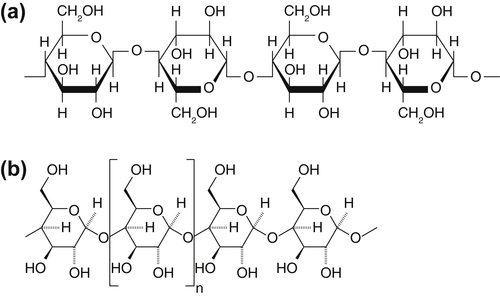
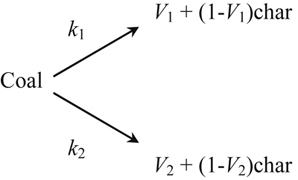
Figure 9.19 shows a characteristic molecular structure of coal, featuring an aromatic carbon backbone and a wide range of bond strengths. General plant matter (as distinct from the fruiting bodies that are often used as food and are primarily composed of starch and sugar) is known as lignocellulosic biomass and is composed of a more or less even mixture of cellulose, hemicellulose, and lignin. As shown in Figure 9.20, cellulose and hemicellulose are both composed of polymers of oxygen-containing ring compounds linked by relatively weak carbon–oxygen bonds. Lignin, in contrast, is composed of small aromatic units connected in weakly linked branched structures. As coal and biomass particles are heated, the internal structure of the carbonaceous material undergoes internal molecular rearrangements. Many weakly bonded moieties break their connecting bonds to the main structure and form gas molecules that aggregate within the solid and, after building sufficient pressure, burst forth from the particle with substantial force. At the same time, some weakly bonded structures and some structures with intermediate-strength bonds pivot about their connecting bond and are able to form stronger, cross-linking bonds with neighboring regions of the structure. Thus there is an inherent competition between solid decomposition and char-forming reactions, with different characteristic activation energies and reaction times. As a consequence, the quantity and chemical composition of the volatile matter that is emitted from these fuels is highly dependent on the nature of the original fuel structure, the rate at which the particles are heated, and the final temperature attained by the particles. For large particles, this also means that the devolatilization process differs as a function of the internal radius of the particle, because the local heating rate varies with the distance from the surface of the particle, where heat is being applied.

The effect of heating rate on evolution of volatiles is most clearly evidenced in the case of woody biomass, which has been shown to have a volatile yield of greater than 90% when small particles are rapidly heated to 1200 °C and to have a volatile yield of only 65% when large particles are slowly heated to 500 °C in the commercial charcoal-making process.
With the importance of the devolatilization process to solid particle combustion and the complexity of the chemical and physical processes involved in devolatilization, a wide variety of models have been developed to describe this process. The simplest models use a single or multiple Arrhenius rates to describe the rate of evolution of volatiles from coal. The single Arrhenius rate model assumes that the devolatilization rate is first order with respect to the volatile matter remaining in the char [40]:
 (9.43)
(9.43)
Fits of Eqn (9.43) to experimental data typically yield an effective activation energy of about 230 kJ/mol, which is consistent with the activation energy for rupturing an ethylene bridge between aromatic rings [41].
The single-rate approach defined by Eqn (9.43) adequately captures the increasing rate of devolatilization at higher temperatures, but fails to account for the observed change in the volatiles yield as a function of temperature. To capture this, Kobayashi et al. [42] proposed the use of two competing reaction paths with different activation energies and different volatile yields (with the higher activation energy path having a higher volatile yield). This “Kobayashi Model” is expressed schematically as
with  ,
, 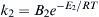 , and V1 and V2 referring to the volatiles yield along each reaction path. With this model, the instantaneous devolatilization rate is the sum of the two independent rates, as shown in Eqn (9.44):
, and V1 and V2 referring to the volatiles yield along each reaction path. With this model, the instantaneous devolatilization rate is the sum of the two independent rates, as shown in Eqn (9.44):
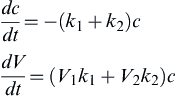 (9.44)
(9.44)
where c refers to the mass of solid coal or char remaining.
Another approach is known as the distributed activation energy model. This model recognizes that devolatilization occurs through many simultaneous reactions. To express this process in a mathematically tractable manner, these reactions are all presumed to be first order and to be describable by a continuous distribution of kinetic rates with a common preexponential and a defined distribution function of activation energy [43]:
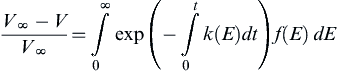 (9.45)
(9.45)
If a Gaussian distribution is used for the activation energy, the distribution function has the form
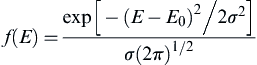 (9.46)
(9.46)
and the distribution of kinetic rates is characterized by a mean (E0) and standard deviation (σ) of the activation energy, so only one additional parameter has been introduced relative to the simplistic approach of using a single Arrhenius rate (Eqn (9.43)).
Advances in the understanding of coal structure and its evolution during devolatilization have led to the development of several coal network models that give predictions of volatile mass evolved as well as predictions of the chemical speciation of the released volatiles [44–46]. Detailed speciation of volatiles is important for understanding NOx and soot formation as well as flame ignition and stability. Extensions of these models to cover biomass fuels have recently been developed, although in some cases the dispersed alkali metal content (primarily potassium) in biomass plays a significant role in catalyzing the reaction steps during devolatilization, making accurate predictions difficult.
The earliest network model came to be known as the functional group depolymerization, vaporization, and cross-linking (FG-DVC) model [44]. The FG portion of the model considers that certain functional groups in the coal lead to the formation of light gas species on heating. The DVC portion of the model describes the deconstruction of the original macromolecular network through bridge-breaking reactions that yield tar and cross-linking reactions that produce char. The FG-DVC model relies heavily on data from thermogravimetric analysis (mass loss during heating) coupled with Fourier transform infrared spectrometer analysis of the evolved gases and uses Monte Carlo simulation of lattice statistics.
A significantly different approach to modeling coal devolatilization is known as chemical percolation devolatilization (CPD) [45]. This approach relies on a detailed description of the chemical structure of the coal, as provided by solid-state nuclear magnetic resonance (NMR) spectroscopy data, which characterizes the fraction of carbon atoms with different types of bonds. The NMR data can be used to infer the average molecular weight of fused aromatic clusters and the number of branching sites for the cluster. In the CPD approach, the NMR data are coupled with percolation statistics to describe the decomposition of the original array of aromatic ring clusters into side chains and reactive intermediates. The side chains convert to light gases and the reactive intermediates undergo a competitive reaction scheme to either form tar or generate light gases and char.
The last of the three major network models is known as FLASHCHAIN [46]. This approach uses a simplified description of the original coal structure as a linear chain, which decomposes according to chain statistics into a broad distribution of fragment sizes. The fragments are partitioned into volatile or condensed species according to mixture-phase equilibrium (i.e., flash distillation). Char formation occurs in the condensed phase with concurrent evolution of fixed gases. Input parameters in the original formulation of this approach included the coal ultimate analysis (i.e., atomic composition), NMR data, and extract yield, but later studies demonstrated that good predictability is still achieved by only using the ultimate analysis and basing the values of the other input parameters on correlation functions.
The ambient pressure has a complex effect on the devolatilization process. For one thing, in most practical situations an increase in ambient pressure will tend to increase the coal particle heating rate for a given reactor temperature. This effect, in and of itself, tends to increase the yield of tar (and therefore of total volatiles), for the reason discussed earlier. However, increasing the ambient pressure also shifts the vapor–liquid equilibrium of the tar species to smaller tar species (with higher vapor pressures) and thus tends to diminish the overall release of tar. Wire-mesh experiments with well-controlled particle heating rates show a significant reduction in the yield of tar and total volatiles as the pressure is increased. The rate of devolatilization, however, is nearly insensitive to pressure, as would be expected for unimolecular reaction processes.
9.5.2. Char Combustion
Once devolatilization of the solid fuel has completed, a porous char particle remains and is consumed through surface reactions of oxidizing species such as O2, H2O, and CO2, as discussed earlier in Section 9.4. The presence of pores in the char particle allows for penetration of reactant species into the particle and therefore for much greater surface area for reactions than is associated with the external surface of the particle. Char particles often have porosities or void fractions of greater than 0.3, depending on the amount of volatiles in the original particle and the extent of particle swelling or shrinkage during devolatilization. Internal surface areas of char particles often exceed 100 m2/g. Using a typical apparent char density of 0.8 g/cm2, one can show that the internal surface area exceeds the external surface area of a particle by over a factor of 10 for a 1 μm particle, by over a factor of 1000 for a 100 μm particle, and by over a factor of 10,000 for a 1 mm particle (the ratio of surface areas scales by particle diameter).
The extent to which a given reactant, such as oxygen, is able to utilize this additional surface area depends on the difficulty in diffusing through the particle to reach the pore surfaces and on the overall balance between diffusion control of the burning rate and kinetic control. To broadly characterize these competing effects, three zones of combustion of porous particles have been identified, as shown in Figure 9.21. In Zone I the combustion rate is fully controlled by the surface reaction rate (kinetically controlled), because the diffusion rate of oxygen is so fast (relatively) that the oxygen concentration throughout the particle and through the particle's boundary layer is essentially equal to the bulk gas oxygen content. Zone I combustion is favored by low temperatures (reducing the surface reaction rate) and small particle sizes (enhancing the surface-specific diffusive flux to the particle). In Zone II the combustion rate is determined by the combined effects of the surface reaction rate and the diffusion of oxygen (both to the particle and through the interior of the particle). The large internal surface area of most char particles coupled with a wide range of pore sizes and different degrees of access to the pores means that this zone is active over a wide range of combustion conditions. In Zone III, the surface burning rate is so fast that oxygen does not effectively penetrate the particle before being consumed. This is diffusion-limited combustion, as was described in Section 9.4. Zone III combustion is favored by high temperatures (increasing the surface burning rate) and large particles (reducing the surface-specific diffusive flux). In one sense, Zone I and Zone III modes of combustion are simply limiting extremes of the general condition of Zone II combustion.

Zone I combustion proceeds at an overall rate equal to the product of the intrinsic burning rate, evaluated at the ambient oxygen concentration, and the total internal surface area. The char diameter necessarily stays constant and the particle density continually decreases as particle mass is evenly removed throughout the particle on the pore surfaces (constant-diameter combustion).
In Zone III combustion, the burning rate is determined by the diffusive flux of oxygen through the particle boundary layer. The particle density remains constant throughout burnout and the particle size continually decreases as mass is removed solely from the particle surface (constant-density combustion).
Zone II combustion proceeds with partial penetration of oxygen, resulting in simultaneous variations in particle density and diameter as the pores closest to the particle surface undergo oxidation, in addition to the external surface of the particle. The ratio of the actual burning rate to the maximum possible rate if the entire particle was subject to the oxygen partial pressure at the external particle surface is known as the effectiveness factor.
9.5.3. Pulverized Coal Char Oxidation
The predominant way in which coal is used to generate steam and electrical power is by means of pulverizing the coal to a fine dust that readily ignites and burns when introduced into a high-temperature furnace. The small particle sizes mean that the coal is readily entrained into the furnace flow and is able to complete combustion before exiting the furnace. Pulverizing the coal also allows a significant portion of its combustion to be completed in the near-burner region, facilitating the use of air-staging and reburning techniques to reduce NOx production. Another significant advantage of pulverized coal combustion relative to other coal combustion techniques, such as fluidized bed combustion, is the ease of scaling boiler sizes to produce upward of 600 MW of electrical power from a single boiler, thereby improving the boiler efficiency and reducing capital costs. Modern pulverized coal boilers can produce electrical power with an overall thermal efficiency of approximately 45%, through the application of a supercritical steam cycle (requiring the use of high-temperature steel alloys in the boiler superheater). Improvements in the manufacture of cost-effective high-temperature steel alloys may improve the thermal efficiency to greater than 50%.
A standard commercial pulverized coal grind usually is required to have 70 wt% of the particles with a size less than 200 mesh (74 μm) and less than 2% over a 50-mesh size (300 μm). With these small particle sizes and the high temperatures within pulverized coal boilers, the coal char particles generally burn in Zone II with combined control of diffusion and chemical reaction. Traditionally the combustion kinetics of pulverized coal have been described through a global particle kinetics approach which calculates the particle reaction rate based on the external surface area of the particle and the concentration of oxygen at the particle surface (thereby accounting for the diffusion profile of oxygen through the particle boundary layer). The dependence of the reaction rate on the partial pressure of oxygen is usually expressed as a power law, yielding a rate expression that is referred to as an “nth-order, Arrhenius” apparent kinetic expression:
![]() (9.47)
(9.47)
where  is equal to the instantaneous burning rate of the particle divided by its external surface area
is equal to the instantaneous burning rate of the particle divided by its external surface area  and is usually expressed in units of kg/(m2 s). According to an analysis of reactions in idealized porous catalysts first reported by Thiele [47] in 1939, in Zone II combustion the apparent activation energy, E, and the apparent reaction order, n, appearing in Eqn (9.47) may be related to the actual, intrinsic kinetic parameters as E = Eint/2 and n = (m + 1)/2, where Eint is the intrinsic activation energy and m is the intrinsic reaction order. The intrinsic activation energy for carbon or char oxidation has been determined to be approximately 180 kJ/mol, and the intrinsic reaction order has been measured (at low to intermediate temperatures) to lie between 0.6 and 1.0 [48]. Smith [49] correlated the oxidation data for a range of carbon types, including porous chars and impervious carbon such as soot to derive the following expression for the intrinsic char oxidation kinetics for an oxygen partial pressure of 101 kPa:
and is usually expressed in units of kg/(m2 s). According to an analysis of reactions in idealized porous catalysts first reported by Thiele [47] in 1939, in Zone II combustion the apparent activation energy, E, and the apparent reaction order, n, appearing in Eqn (9.47) may be related to the actual, intrinsic kinetic parameters as E = Eint/2 and n = (m + 1)/2, where Eint is the intrinsic activation energy and m is the intrinsic reaction order. The intrinsic activation energy for carbon or char oxidation has been determined to be approximately 180 kJ/mol, and the intrinsic reaction order has been measured (at low to intermediate temperatures) to lie between 0.6 and 1.0 [48]. Smith [49] correlated the oxidation data for a range of carbon types, including porous chars and impervious carbon such as soot to derive the following expression for the intrinsic char oxidation kinetics for an oxygen partial pressure of 101 kPa:
![]() (9.48)
(9.48)
Because of the variability in deduced reaction orders for different experiments and carbon types, a general expression for the kinetic rate that includes the oxygen dependence could not be determined.
In reality, it is believed that the oxidation of carbonaceous surfaces occurs through adsorption of oxygen, either immediately releasing a carbon monoxide or carbon dioxide molecule or forming a stable surface oxygen complex that may later desorb as CO or CO2. Various multistep reaction schemes have been formulated to describe this process, but the experimental and theoretical information available to date has been insufficient to specify any surface oxidation mechanism and associated set of rate parameters with any degree of confidence. As an example, Mitchell et al. [50] has proposed the following surface reaction mechanism:

where an O or O2 in parentheses denotes a surface oxide, Cf represents a surface carbon atom with an open bond, and Cb represents a “bulk” carbon atom (i.e., one with fully assigned bonds).
9.5.4. Gasification and Oxycombustion
Fuel-rich oxidation of solid carbonaceous fuels to form gaseous fuel components such as methane, hydrogen, and carbon monoxide is known as gasification. Coal gasification has been an industrially important technology for many decades and continues to be used for the generation of synthetic natural gas, hydrogen, ammonia, and specialty chemicals. Recently, gasification of coal has arisen as an important technology for high efficiency generation of electrical power using large aeroderivative gas turbine engines with a bottoming steam power cycle. This technology, known as integrated gasification combined cycle (IGCC), can currently achieve overall thermal efficiencies near 45% with very low emissions of traditional pollutants such as NOx, SO2, and mercury. In addition, by converting the carbon monoxide in the gasifier product gas to carbon dioxide and hydrogen according to the water–gas shift reaction,
![]()
gasification of coal and biomass can yield large quantities of relatively inexpensive hydrogen and readily allow for capture and subsequent sequestration of the fuel carbon (in the form of CO2). A typical system layout for coal or biomass gasification to produce hydrogen and electricity while capturing carbon is shown in Figure 9.22. Cogasification of coal and biomass with carbon capture and sequestration is a promising technology for power and/or fuel production that is not merely carbon neutral but can be carbon negative. Gasification of solid fuels also allows for conversion of the solid fuel feedstock into one or more liquid fuels by processing the gasifier syngas (cleaned of impurities such as H2S, HCN, and NH3) over catalytic reactors to yield methanol, ethanol, or even gasoline and diesel fuel, through the Fischer–Tropsch catalytic synthesis process originally developed in Germany.
Analogous to solid fuel boilers, gasifiers can be operated either as fluidized bed devices with relatively large fuel particles, moderate temperatures, and long particle residence times, or as entrained flow devices with pulverized fuel particles, high temperatures, and short residence times. Either air or oxygen can be used as the primary oxidant gas, although steam is generally added to assist in char gasification, according to the following reaction:

![]()
In addition to char gasification from steam, CO2 produced locally from consumption of oxygen in the gasifier reacts with char according to the Boudouard reaction (previously shown). The generation of CO and CO2 from the oxygen consumed in the gasifier releases the energy required to devolatilize the raw fuel particles and gasify the resultant char through the endothermic reactions with steam and CO2.
Air-blown gasifiers benefit from a low-cost (free) source of oxidant, but produce a gasifier product gas that has a very low heating value (approximately 5 MJ/m3 for air-blown gasification of coal, compared to 38 MJ/m3 for a typical natural gas) and which is too dilute to use for liquid fuel synthesis. Oxygen-blown gasifiers yield a moderate heating value product gas (approximately 9 MJ/m3) and can operate at very high temperatures, but suffer from the significant cost of producing oxygen with an air-separation unit.
Because CO2 and H2O react much slower with char than O2 reacts with char, gasifiers usually operate at elevated pressure (to increase the collision rate of the gasifying species with the char). For IGCC systems, operating the gasifier at elevated pressure eliminates the need for additional compression before injection of the product gases into the gas turbine combustor. Biomass gasifiers are typically operated at atmospheric pressure because of the difficulty of feeding biomass particles into a pressurized gasifier and because the high biomass volatile content and high reactivity of biomass chars obviate some of the need for elevated pressures to obtain good carbon conversion.
An alternative technology to gasification for power production with carbon capture and sequestration is to burn the solid fuel in oxygen, instead of air. To moderate the combustion temperatures and avoid the formation of molten, slagging deposits of mineral matter on the boiler tubes, the oxygen is usually diluted with recycled flue gas before entering the combustion chamber. The very high concentration of carbon dioxide in the exhaust gas from this process allows fairly simple capture of CO2 (the major additional financial cost and efficiency loss in this system result from the oxygen generation process). In addition, compression of the flue gas for CO2 transport allows capture of pollutant species, making this a near-zero emission process. In contrast to the option of installing new gasification power plants, the oxy-fuel combustion technology is generally believed to be retrofitable to existing boiler systems, thereby taking advantage of the large capital investment that has already been made in these boilers.
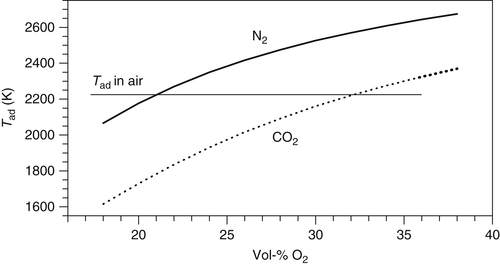
A critical consideration in oxy-fuel combustion systems is the extent of flue gas recirculation that is desired. From a cost perspective, one would like to minimize the amount of recirculation, thereby increasing the oxygen concentration of the gases entering the boiler. However, the volatile flame temperature and char combustion temperature both increase substantially with higher oxygen content. Counteracting this influence of increasing oxygen content is the substitution of CO2 for N2 as the predominant gas in the boiler. Carbon dioxide has a molar specific heat that is 1.7 times larger than that of molecular nitrogen. Therefore, combustion in CO2 environments results in lower flame temperatures than combustion in equivalent N2 environments. Figure 9.23 shows the computed adiabatic flame temperature for stoichiometric combustion of methane in mixtures of oxygen with nitrogen and oxygen with carbon dioxide. As is evident in this figure, combustion of methane in 32% O2 in CO2 produces the same flame temperature as combustion in dry air (with 21% O2 in N2). A further, important consideration for boiler operation is the thermal radiation emitted by hot CO2 in the furnace—in contrast to N2, CO2 emits thermal radiation in the infrared and can impact the balance between radiant and convective heat transfer in a boiler. In addition, the flue gas recycle substantially increases the water vapor content of the furnace gases, thereby enhancing thermal radiation from hot H2O.
For application to combustion of solid fuels, the presence of high concentrations of CO2 decreases the volatile flame temperature (for a given O2 level) because of the higher specific heat of CO2. Also, CO2 decreases the volatile combustion rate because of the reduced diffusivity of O2 through CO2 atmospheres (the diffusivity is about 20% lower). For char combustion, high levels of CO2 decrease the burning rate during Zone II and Zone III combustion because of the lower oxygen diffusivity. It is possible that CO2 enhances the overall kinetic rate of carbon oxidation through contributions from the Boudouard reaction, but it is generally believed that this reaction is too slow to effectively compete with char oxidation from O2, even at the very high particle temperatures that are sometimes experienced with the use of high oxygen concentrations. NOx formation, both from oxidation of nitrogen in the volatile fuel components and from oxidation of the nitrogen in the char, is enhanced for the high volatile flame temperatures and elevated char combustion temperatures associated with enhanced oxygen levels. However, practical oxy-combustion systems recycle a substantial fraction (typically on the order of 70%) of the flue gas back to the combustion furnace, allowing significant reburning of the previously formed NOx in the flame zone and on the burning char particles. Also, substitution of CO2 for N2 eliminates the contribution of thermal NOx formation. As a result, the net NOx production is reduced for oxy-fuel combustion processes, typically by a factor of three or more.
Another approach to oxy-fuel combustion for carbon capture and sequestration is chemical looping combustion [53,54]. Chemical looping combustion makes use of an oxygen carrier, such as a metal oxide, to transport oxygen to the fuel, thus preventing air from ever being in direct contact with the fuel during combustion. The process is conducted with two reactors, a reducer (or fuel reactor) and an oxidizer (or air reactor) as illustrated in Figure 9.24. In the reducer, coal (or fuel) reacts with particulate metal oxide to form CO2, H2O, and particulate metal (or a metal oxide with a lower oxidation state). Depending on the metal oxide, this process is overall endothermic or slightly exothermic. The water formed results from oxidation of volatiles originating from the coal (or from an enhancer gas introduced to assist the reaction). In the oxidizer (or air reactor), the metal particles are reacted with air to reform the metal oxide through an exothermic reaction that may be used to supply energy to a heat engine. Low grade heat rejected from the heat engine is returned to the fuel reactor as the distribution of heat release from the two reactors is such that the temperature of the air reactor is operated at a higher temperature than that of the fuel reactor. The particulate metal oxide and metal are transported back and forth between reactors. Carbon dioxide capture is possible because the gaseous products of the fuel reactor consist largely of CO2 and H2O, and the air reactor is devoid of carbon.

The reactions in the air reactor are those described earlier in this chapter for metal combustion and can involve the two overall steps
![]()
![]()
where M refers to the metal and MO the metal oxide. Note the first step forms only thermal energy and the latter provides both thermal energy and a source of hydrogen. In the fuel reactor, the reduction process has been proposed to occur through several possible mechanisms depending on the metal oxide and the operating conditions of the reducer, particularly the temperature. Indirect reduction may occur with the aid of gaseous intermediates according to
![]()
![]()
With coal as the fuel, these reactions may be initiated through devolatilization
![]()
in which hydrogen may also reduce the metal oxide to form water vapor. An alternative process, which has been referred to as chemical looping with oxygen uncoupling, involves forming a gaseous oxidizer through the decomposition of the metal oxide.

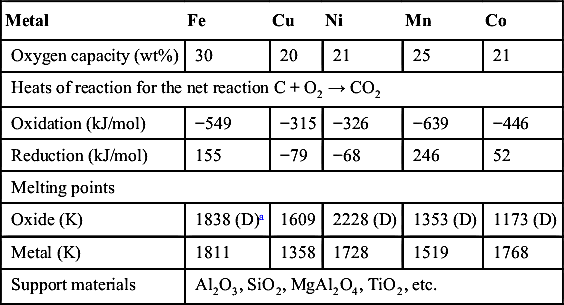
Table 9.9
Metals for Chemical Looping Combustion
| Metal | Fe | Cu | Ni | Mn | Co |
| Oxygen capacity (wt%) | 30 | 20 | 21 | 25 | 21 |
| Heats of reaction for the net reaction C + O2 → CO2 | |||||
| Oxidation (kJ/mol) | −549 | −315 | −326 | −639 | −446 |
| Reduction (kJ/mol) | 155 | −79 | −68 | 246 | 52 |
| Melting points | |||||
| Oxide (K) | 1838 (D)a | 1609 | 2228 (D) | 1353 (D) | 1173 (D) |
| Metal (K) | 1811 | 1358 | 1728 | 1519 | 1768 |
| Support materials | Al2O3, SiO2, MgAl2O4, TiO2, etc. | ||||
At temperatures below the decomposition temperature of the metal oxide, direct solid–solid reactions have also been proposed [55].
![]()
For complete oxidation, the stoichiometry of the carbon reaction with oxygen and the metal oxide should be written to form CO2. Table 9.9 lists the primary metals that have been considered, their oxygen capacity, and the theoretical heat release distribution between the two reactors assuming an overall process reaction of C + O2 → CO2. Many other factors are important to the choice of the oxygen carrier including particle kinetics, support materials, strength, attrition, environmental and health concerns, and cost [54].
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.