
9.6. Soot Oxidation (C. R. Shaddix)
A detailed description of the chemical processes involved in soot formation was given in the previous chapter. Oxidation of soot particles is an important subject area because in most practical processes that form soot (e.g., diesel engines, gas turbines, and boilers) much more soot is formed in the flames than is emitted as a pollutant. In other words, it is the incomplete oxidation of soot within flames that leads to soot emissions. Also, the oxidation rate of soot controls the amount of soot that exists in the highest temperature regions of a flame and thereby has a large influence on the radiant energy emitted from flames. Most boiler and furnace systems (especially glass melting furnaces) rely on thermal radiation from hot, oxidizing soot to transfer heat effectively to the walls or other heat-absorbing surface.

As carbonaceous particles are composed primarily of carbon, the chemical oxidation of soot proceeds in a similar manner to that previously described for char particles. However, soot particles in combustion processes are generally composed of small, isolated spheres, typically on the order of 30 nm in diameter, or, more generally, as open-structured aggregates of such spheres, as shown in Figure 9.25. While aggregated particles may reach very large characteristic sizes (up to or even larger than 1 μm), the open structure of most aggregates allows gas molecules to freely diffuse to each individual sphere (known as a primary particle). As a result, the characteristic particle size for oxidation is on the order of the primary particle size, such that under most conditions the burning rate is kinetically controlled (Zone I combustion) and a single-film model of the particle combustion process is appropriate (i.e., the residence time of CO through the boundary layer is too short to allow oxidation to CO2).
Soot can be oxidized by molecular oxygen, through the process described previously for pulverized coal char oxidation, but in combustion systems the dominant oxidation of soot is performed by radical species, especially the hydroxyl radical, OH. This discovery was made by Fenimore and Jones [56] using a two-stage burner system in which the fuel-rich products (including soot) from a premixed flat flame were cooled and mixed with oxygen, hydrogen, and diluent gas before being burned in a second flat flame that was operated at stoichiometries ranging from fuel lean to slightly fuel rich. The importance of soot oxidation by OH was later confirmed by several investigators using both two-stage burners [57] and laminar diffusion flames [58–60]. Oxygen atoms (O) contribute to soot oxidation, but their concentrations are always lower than that of OH in flames, so OH tends to dominate the oxidation process.
The reaction probabilities for O and OH with soot particles have been measured by Roth and coworkers in a series of shock tube experiments [61–63]. They have found that both radicals react with soot particles with a collision efficiency of between 0.10 and 0.20. In contrast, the reaction probability with O2 is at least an order of magnitude lower [58]. Of course, at lower temperatures and sufficiently lean mixtures, soot oxidation by radical species becomes small and oxidation by O2 is important (though slow). Consequently, soot that passes through or avoids the primary reaction zone of a flame (e.g., due to local flame quenching) may experience oxidation from O2 in the postflame gases. Analysis of soot oxidation rates in flames [57–60] has supported the approximate value of the OH collision efficiency determined by Roth and coworkers.
Unfortunately, OH and O concentrations in flames are determined by detailed chemical kinetics and cannot be accurately predicted from simple equilibrium at the local temperature and stoichiometry. This is particularly true when active soot oxidation is occurring and the local temperature is decreasing with flame residence time [62]. As a consequence, most attempts to model soot oxidation in flames have by necessity used a relation based on oxidation by O2 and then applied a correction factor to augment the rate to approximate the effect of oxidation by radicals. The two most commonly applied rate equations for soot oxidation by O2 are those developed by Lee et al. [64] and Nagle and Strickland-Constable [65].
Lee et al. were able to fit their data of soot burnout at the top of a flame by employing a simple first-order rate equation:
![]() (9.49)
(9.49)
where 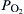 is given in atm, T is in Kelvin, and R has the units of kJ/(mol K). This may be further simplified to pure Arrhenius form as
is given in atm, T is in Kelvin, and R has the units of kJ/(mol K). This may be further simplified to pure Arrhenius form as
![]() (9.50)
(9.50)
Comparison of the Lee rate with the rate derived by Smith for a variety of carbon types (Eqn (9.48)) shows that the Lee rate is twice as high as the Smith rate at 1800 K.
Nagle and Strickland-Constable performed oxidation measurements on pyrocarbon rods subjected to a high velocity oxidizer. For temperatures greater than 2000 K they measured a dip in the oxidation rate until the surface reached a temperature that was substantially greater and the oxidation rate once again increased with increasing temperature. To explain this phenomenon, they proposed that carbon oxidation proceeds through two types of surface sites, types A and B, that both react with oxygen to produce CO and generate a new type A site. Type A sites, meanwhile, thermally rearrange to type B sites. These presumptions yield an overall rate equation of the form
![]() (9.51)
(9.51)
where
![]()
With appropriate choices of kinetic constants kA, kB, kT, and kZ, this approach can reproduce the Nagle and Strickland-Constable experimental data quite well [66]. Park and Appleton [66] oxidized carbon black particles in a series of shock tube experiments and found a similar dependence of oxidation rate on oxygen concentration and temperature as that of Nagle and Strickland-Constable. Of course, the proper kinetic approach for soot oxidation by O2 undoubtedly should involve a complex surface reaction mechanism with distinct adsorption and desorption steps, in addition to site rearrangements, as suggested previously for char surface combustion.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.