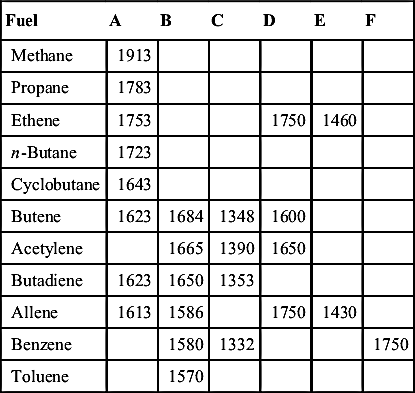In earlier sections of this chapter, the role that particulates play in a given environmental scenario was identified. This section will be devoted exclusively to combustion-generated particulates whose main constituent is carbon. Those carbonaceous particulates that form from gas-phase processes are generally referred to as soot, and those that develop from pyrolysis of liquid hydrocarbon fuels are generally referred to as coke or cenospheres.
Although various restrictions have been placed on carbon particulate emissions from different types of power plants, these particles can play a beneficial as well as detrimental role in the overall plant process. The detrimental effects are well known. The presence of particulates in gas turbines can severely affect the lifetime of the blades; soot particulates in diesel engines absorb carcinogenic materials, thereby posing a health hazard. It has even been postulated that after a nuclear blast, the subsequent fires would create enormous amounts of soot whose dispersal into the atmosphere would absorb enough of the sun’s radiation to create a “nuclear winter” on Earth. Nevertheless, particulates can be useful. In many industrial furnaces, for example, the presence of carbon particulates increases the radiative power of the flame, and thus can increase appreciably the heat transfer rates.
The last point is worth considering in more detail. Most hydrocarbon diffusion flames are luminous, and this luminosity is due to carbon particulates that radiate strongly at the high combustion gas temperatures. As discussed in Chapter 6, most flames appear yellow when there is particulate formation. The solid-phase particulate cloud has a high emissivity compared with a pure gaseous system; thus, soot-laden flames appreciably increase the radiant heat transfer. In fact, some systems can approach black-body conditions. Thus, when the rate of heat transfer from the combustion gases to some surface, such as a melt, is important—as is the case in certain industrial furnaces—it is beneficial to operate the system in a particular diffusion flame mode to ensure formation of carbon particles. Such particles can later be burned off with additional air to meet emission standards. But some flames are not as luminous as others. Under certain conditions the small particles that form are oxidized in the flame front and do not create a particulate cloud.
It is well known that the extent of soot formation is related to the type of flame existing in a given process. Diesel exhausts are known to be smokier than those of spark-ignition engines. Diffusion flame conditions prevail in fuel-injected diesel engines, but carbureted spark-ignition engines entail the combustion of nearly homogeneous premixed fuel–air systems.
The various phenomena involved in carbon particulate formation have been extensively studied. The literature is abundant and some extensive review articles [53–55] are available. Most of the subsequent material in this chapter will deal with soot formation while a brief commentary on the coke-like formation from liquid fuels will be given at the end.
8.5.1. Characteristics of Soot
The characteristics of soot are well described in the article by Palmer and Cullis [53], who provide detailed references on the topic. Aspects of their review are worth summarizing directly. They report the detailed physical characteristics of soot as follows:
The carbon formed in flames generally contains at least 1% by weight of hydrogen. On an atomic basis this represents quite a considerable proportion of this element and corresponds approximately to an empirical formula of C8H. When examined under the electron microscope, the deposited carbon appears to consist of a number of roughly spherical particles, strung together rather like pearls on a necklace. The diameters of these particles vary from 100 to 2000 Å and most commonly lie between 100 and 500 Å. The smallest particles are found in luminous but nonsooting flames, while the largest are obtained in heavily sooting flames. X-ray diffraction shows that each particle is made up of a large number (104) of crystallites. Each crystallite is shown by electron diffraction to consist of 5–10 sheets of carbon atoms (of the basic type existing in ideal graphite), each containing about 100 carbon atoms and thus having length and breadth of the order of 20–30 Å. But the layer planes, although parallel to one another and at the same distance apart, have a turbostratic structure, that is, they are randomly stacked in relation to one another, with the result that the interlayer spacing (3.44 Å) is considerably greater than in ideal graphite (3.35 Å). It may readily be calculated on this picture of dispersed carbon deposits that an “average” spherical particle contains from 105 to 106 carbon atoms.
Investigators have used the words “carbon” and “soot” to describe a wide variety of carbonaceous solid materials, many of which contain appreciable amounts of hydrogen as well as other elements and compounds that may have been present in the original hydrocarbon fuel. The properties of the solids change markedly with the conditions of formation; and, indeed, several well-defined varieties of solid carbon may be distinguished. One of the most obvious and important differences depends on how the carbon is formed: Carbon may be formed by a homogeneous vapor-phase reaction; it may be deposited on a solid surface that is present in or near the reaction zone; or it may be generated by a liquid-phase pyrolysis.
8.5.2. Soot Formation Processes
Determining the relative tendency of hydrocarbon fuels to soot, explaining why this relative tendency between fuels exists, and discovering how to control or limit soot production in a particular combustion process—these are the elements of greatest importance to the practicing engineer. Since the amount of soot formed from a particular fuel has a complex dependence on the overall combustion process, no single characteristic parameter can define the amount formed per unit weight of fuel consumed. Both the flame type and various physical parameters determine the extent of soot formation from a given fuel. Moreover, depending upon the combustion process, the final mass of particulates emitted from the system could be reduced by a particle afterburning process.
Examining the detailed chemical processes of soot formation and oxidation, one notes how complex the overall system is. Regardless of the flame type, the fuel undergoes either pure or oxidative pyrolysis. If the fuel is nonaromatic, some precursors of the fuel pyrolysis undergo cyclization to create an aromatic ring. The ring structure is thought to add alkyl groups, developing into a polycyclic aromatic (PAH) structure that grows owing to the presence of acetylene and other vapor-phase soot precursors. Under certain flame conditions, the elements forming large aromatic structures are simultaneously oxidized. The precursors and all subsequent structures must be conjugated so that they are resonance-stabilized at the high temperatures in which they grow. Eventually, the aromatic structures reach a large enough size to develop into particle nuclei. Such condensed-phase carbon particles contain large amounts of hydrogen. The particles dehydrogenate (age) in a high-temperature combustion field while physically and chemically absorbing gaseous hydrocarbon species which also dehydrogenate, thereby effecting a large increase of soot mass. The growing particles also agglomerate and conglomerate. To a large degree, the absorbed species undergo chemical reformation, which results in a carbonaceous soot structure. While these events are occurring, oxidative attack on the particles continues to form gaseous products. Nevertheless, the eventual chemical steps in the soot formation process appear to be similar, regardless of the initial fuel. As Palmer and Cullis [53] noted, “With diffusion flames and premixed flames investigations have been made of the properties of the carbon formed and of the extent of carbon formation under various conditions. In general, however, the properties of the carbon formed in flames are remarkably little affected by the type of flame, the nature of the fuel being burnt and the other conditions under which they are produced.”
Although many current investigations have shown that the physical and chemical processes noted above are both complex and various, the generalized model to be described below allows one to identify certain controlling steps, thus gaining some important insights into the relative tendency of fuels to soot under particular flame configurations. Using this insight, one may develop means to control the amount of particulate emission.
8.5.3. Experimental Systems and Soot Formation
Estimates of fuel sooting tendency have been made using various types of flames and chemical systems. In the context to be used here, the term “sooting tendency” generally refers to a qualitative or quantitative measure of the incipient soot particle formation and growth rates of one fuel relative to another in a particular experimental combustion configuration. In many cases, this tendency varies strongly with the type of flame or combustion process under investigation. This variation is important because the incipient soot particle formation and growth rates determine the soot volume fraction formed and emitted in a combustion system.
For premixed fuel−air systems, results are reported in various terms that can be related to a critical equivalence ratio at which the onset of some yellow flame luminosity is observed. Premixed combustion studies have been performed primarily with Bunsen-type flames [56,57], flat flames [58], and stirred reactors [59,60]. The earliest work [61,62] on diffusion flames dealt mainly with axisymmetric coflow (coannular) systems in which the smoke height or the volumetric or mass flow rate of the fuel at this height was used as the correlating parameter. The smoke height is considered to be a measure of the fuel’s particulate formation and growth rates but is controlled by the soot particle burnup. The specific references to this early work and that mentioned in subsequent paragraphs can be found in Ref. [54].
Work on coflowing Wolfhard−Parker burners [63,64], axisymmetric inverse coflowing configurations (oxidizer is the central jet) [65,66], and counterflow (opposed-jet) diffusion flames [67–69] has employed chemical sampling and laser diagnostics for scattering, extinction, and fluorescence measurements to determine chemical precursors, soot number density, volume fraction, and average particle size. These experiments not only obtained a measurement of where and when incipient soot formation takes place in a particular flame configuration, but also followed the variation of the number density, volume fraction, and size with time after incipient formation. An important feature of most of these flame configurations is that they separate any particle burnup from the formation rate. As described in Chapter 4, counterflow experiments are designed to be directly opposing gas jets; a flow stream, normally the oxidizer, opposing the fuel flow emanating from a horizontal porous cylinder; or a directly opposing jet emanating from a cylindrical tube. Thus, the porous cylinder condition creates flames that emulate the wakes of burning liquid droplets in various types of streams. A small liquid droplet burning in perfect spherical symmetry is nonluminous; larger droplets, because of a longer diffusion time from the liquid surface to the flame front, facilitate fuel pyrolysis and growth and have mildly luminous flames. In another approach, shock tube experiments in which fuel pyrolysis leads to soot also provide some means of measuring the relative sooting tendency, but not necessarily applicable to a combustion process. Confusion can arise, however, in interpreting soot formation results unless one understands how the structures of the processes taking place in premixed flames, normal coflowing, inverse coflowing, and counterflowing diffusion flames; or shock tubes differ. This point cannot be overemphasized.
In premixed flames, the formation of soot precursors through fuel pyrolysis is competitive with oxidative attack on these precursors, but no oxidative attack occurs on the precursors formed during the fuel pyrolysis in diffusion flames. However, in normal coflowing diffusion flames, the fuel stream, its pyrolysis products, and other constituents added to it are convected by fluid motion across the flame front; that is, convection dominates molecular diffusion. These constituents flow into an environment of increasing temperature. Recall that, for a pure fuel jet (a mass fraction of 1) in counterflow diffusion flames, all elements in the fuel stream reach the flame front only by molecular diffusion across the stagnation plane. Soot particles form only on the oxidizer side of the flame front in a pure air−fuel system; then they are convected toward the stagnation plane and are not oxidized. Unfortunately, this counterflow procedure does not occur in any industrial combustion process. The flow pattern in inverse axisymmetric coflowing flames is such that the precursors and particulates formed are also convected away from the flame zone. In shock tubes, fuel pyrolysis (and soot formation) takes place at a relatively fixed temperature and pressure. Thus, it is possible to measure a relative sooting parameter at a fixed temperature. Although stoichiometric temperatures similar to the temperatures reached in shock tubes can be established with coflowing diffusion flames, soot formation in these flames occurs at much lower temperatures prior to reaching the stoichiometric flame surface. In diffusion flames, the temperature–time history of a fuel element leading to pyrolysis, particulate formation, and growth varies; in shock tube experiments it generally does not.
Flame turbulence should not affect soot formation processes under premixed combustion conditions, and the near correspondence of the results from Bunsen flames [56] and stirred reactors [59] tends to support this contention.
8.5.4. Sooting Tendencies
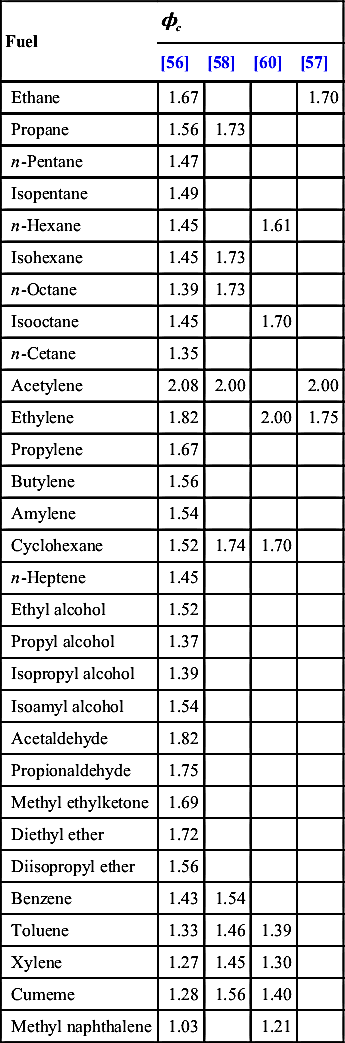
Most of the early work and more recent efforts on soot formation under premixed conditions were conducted with air as the oxidizer. These early results reported data as a critical sooting equivalence ratio ϕc, in which the higher the ϕc, the lower the tendency to soot. As shown in Table 8.5, the trend observed followed the order
Aromatics>Alkanes>Alkenes>Alkynes
that is, ethane was reported to have a greater sooting tendency than ethene or acetylene.
The most extensive early data of sooting under laminar diffusion flame conditions, as measured by the smoke height method, were obtained by Schalla et al. [61,62]. The general trend observed followed the order
Aromatics>Alkynes>Alkenes>Alkanes
Upon comparison, the trends of the alkyl compounds in the premixed case revealed an inconsistency with the oxidation reaction mechanism of ethene under fuel-rich conditions: it was found that the alkane was rapidly converted first to the alkene and then to the alkyne [70,71]. Thus one would expect the alkynes to exhibit the greatest sooting propensity under premixed conditions. The answer to this inconsistency was developed from the early work of Milliken [72], who studied the soot emission from ethene on a cooled flat flame burner. Milliken found that the cooler the flame, the greater the tendency to soot (i.e., the lower the critical sooting equivalence ratio). He explained this temperature trend by evaluating the effect of temperature on the two competing processes occurring in the sooting flame: the pyrolysis rate of the fuel intermediate (acetylene precursor) leading to the other precursors and the rate of oxidative (hydroxyl radical, OH) attack on these precursors. Both rates increase with temperature, but the oxidative rate increases faster. Thus the tendency of premixed flames to soot diminishes as the temperature rises. Other investigators [71,73,74] verified Milliken's calculations and extended the concept by showing that in coflow diffusion flames, where there is no oxidative attack on the precursors, the soot volume fraction created increases with temperature. Therefore, for a proper comparison of the effect of fuel structure on the sooting tendency, the flame temperature must be controlled. As will be noted subsequently, the temperature field created by the diffusion flame is more important than the actual flame temperature [75].
Table 8.5
Critical Sooting Equivalence Ratios (ϕc) of Various Fuels Premixed with Air
Early work on premixed Bunsen flames and coflowing diffusion flames [73-76] was repeated in experiments where the temperature was controlled by varying the N2/O2 ratio in the oxidizer used for the Bunsen experiments and by adding inerts, particularly nitrogen, to the fuel in the diffusion flame experiments. The critical sooting equivalence ratio in premixed flames was determined by the visual observation of the onset of soot luminosity. Varying the nitrogen concentration permitted the determination of a critical sooting equivalence ratio ψc (whereas ϕc is based on a stoichiometry referenced to carbon dioxide and water as products; ψc is based on a stoichiometry referenced to carbon monoxide and water as products since all conditions of operation are fuel rich) as a function of the calculated adiabatic flame temperature. The results are shown in Figure 8.13. The values corresponding to air agree exactly with the early work of Street and Thomas [56]. However, at a fixed temperature, ψc (acetylene) < ψc (ethene) < ψc (ethane), as one would expect. Also, there is no specific trend with respect to homologous series.
Figure 8.13Critical sooting equivalence ratios (ψc) based on CO and H2O stoichiometry of various hydrocarbon fuels as a function of calculated adiabatic flame temperature Tf.
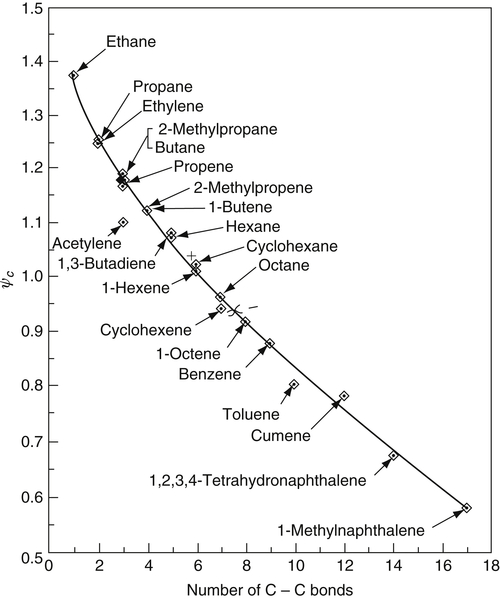
Following the conceptual idea introduced by Milliken [73], Takahashi and Glassman [57] showed, with appropriate assumptions, that, at a fixed temperature, ψc could correlate with the “number of C–C bonds” in the fuel and that a plot of the log ψc versus number of C–C bonds should give a straight line. This parameter, number of C–C bonds, serves as a measure of both the size of the fuel molecule and the C/H ratio. In pyrolysis, since the activation energies of hydrocarbon fuels vary only slightly, molecular size increases the radical pool size. This increase can be regarded as an increase in the Arrhenius pre-exponential factor for the overall rate coefficient and hence in the pyrolysis and precursor formation rates so that the C/H ratio determines the OH concentration [77]. The ψc versus C–C bond plot is shown in Figure 8.14. When these data are plotted as log ψc versus C–C bonds, a straight line is obtained [57]. Also plotted on Figure 8.14, as a test of the correlation, are the results of some fuel mixtures [78]. These data lead to the important conclusion that specific fuel structures do not play a role under premixed conditions; for example, decane has the same sooting tendency as benzene at the same premixed flame temperature. Thus, under premixed conditions, all fuels must break down to the same essential species that build into a soot particle. This postulate is consistent with the results of Harris and Weiner [79,80], who showed that increments of carbon introduced as ethene in premixed flames were just as effective in producing soot as increments of carbon introduced as toluene and that the greatest mass of soot came about by growth on particles in which the dominant species contributing to this particle growth was acetylene.
Figure 8.14Critical sooting equivalence ratio ψc at 2200 K as a function of the “number C–C bonds” in hydrocarbon fuels. +, 0, and − indicate ethene/1-octene mixtures in molar ratios of 5 to 1, 2 to 1, and 1 to 2, respectively; χ, acetylene/benzene at a molar ratio of 1 to 3. The O symbol for 2 to 1 falls on top of the butene symbol.
Similar temperature-control procedures were applied to a highly overventilated concentric coflowing diffusion flame [55,81] in which 29 fuels were evaluated with respect to their sooting tendency. In these experiments the mass flow rate of the fuel jet is increased until soot breaks through the top of the “luminous visible flame front.” This luminous visible flame front is not the flame; its edge defines the region in which the soot particles formed were completely burned. As stated earlier the stoichiometric (Burke-Schumann) flame front that defines the flame temperature lies within this luminous envelope. The initial breakthrough of this luminous boundary is referred to as the sooting or smoke height, and most data are reported in terms of the fuel mass flow rate at this height. Indeed the initial breakthrough does not occur at the fuel jet axis, but in a toroidial ring around the axis. A planar view of a smoke height condition appears as two wings. This ring develops because the flow streamlines in the axisymmetric coflow approach are not parallel.
If nitrogen, or any inert species, is added to the fuel jet when the smoke height is reached, the luminous zone closes and soot no longer emanates from the top of the flame. If fuel mass flow is then increased, another smoke height is reached. Additional inert diluent again suppresses the soot emanation and additional fuel flow is necessary for the flame to smoke. In this manner the smoke height, or mass flow rate at the smoke height, of a single fuel may be obtained at different flame temperatures since the diluent lowers the temperature. Whether the effect of diluent on soot exhaust through a flame front is due mainly to temperature variation is also discussed subsequently.
How this smoke effect varies with inert addition is best explained by considering the results of many early investigators who reported that incipient soot formation occurred in a narrow temperature range. The various results are shown in Table 8.6. Since, as stated earlier, the incipient particle formation mechanisms for various fuels follow similar routes, it seems appropriate to conclude that a high activation energy process or processes control the incipient particle formation. The best concept and evidence to date are those given by Dobbins [82]. It is likely that the slight variation of temperatures shown in Table 8.6 is attributable to the different experimental procedures used. The experimental procedure of Ref. [83] sheds some light on the concept proposed here. In these experiments, a given sooting flame was diluted until all particle radiation just disappeared, then the temperature of the flame front was measured on the centerline of the coannular configuration. These measurements are shown in Figures 8.15 and 8.16. Since different amounts of dilution were required to eliminate particle continuum radiation for the different fuels, it should not be surprising that there is some variation for different fuels in the temperature limit for incipient particle formation.
Figure 8.15Typical temperature profiles along the centerline of laminar hydrocarbon fuel jets diluted with N2 to the point of no luminosity when burning in overventilated air streams. H is the height of the flame; z is the distance from jet exit along the centerline.
Figure 8.16Incipient soot formation temperature as a function of the amount of diluent. The ordinate is the temperature in kelvins; the abscissa is f = Qf/(Qf + Qdg) × 100, where Qf is the volumetric flow rate of the fuel and Qdg is that of the diluent.
Table 8.6
Soot Inception Temperature (K) by Various Investigators
Fuel
A
B
C
D
E
F
Methane
1913
Propane
1783
Ethene
1753
1750
1460
n-Butane
1723
Cyclobutane
1643
Butene
1623
1684
1348
1600
Acetylene
1665
1390
1650
Butadiene
1623
1650
1353
Allene
1613
1586
1750
1430
Benzene
1580
1332
1750
Toluene
1570
A. Dilution of fuel jet until no luminosity (Smith, C. A., M.S. E. Thesis, Dep. Mech. Aerosp. Eng., Princeton Univ., Princeton, New Jersey, 1990).
B. Dilution of fuel jet until no luminosity (Glassman et al. [83]).
C. Centerline observation of first particulates (Gomez et al. [98]).
D. Optical measurements of inception and temperature (Boedecker, L. R., and Dobbs, G. M., Proc. Combust. Instit.21, 1097, 1986).
E. Critical sooting C/O ratio in premixed flames as a function of controlled flame temperature (Bohm, H., Hesse, D., Jander, H., Luers, B., Pietscher, J., Wagner, H. Gg., and Weiss, M., Proc. Combust. Instit. 22, 403, 1988).
F. Flat premixed flame observations and measurements (Olson, D. B., and Madronvich, S., Combust. Flame60, 203, (1985)).
If the temperature limit is taken as the average of those reported in Figures 8.15 and 8.16 (∼1650 K), it can be assumed that incipient particle formation occurs along a 1650 K isotherm in the coannular diffusion flame structure. Then, at the initial smoke height with no inert addition, the 1650 K isotherm within the fuel flow designates the point of incipient particle formation. Thus, there is enough growth along flow streamlines that coincides with and closely parallels the 1650 K isotherm to cause particles to penetrate the flame and smoke. This flow coincidence will, of course, be near the sides of the flame. For this reason, the smoke height is observed as a flame breakthrough, lending a wing-like appearance to the flame luminous envelope [55]. Thus, what determines the smoke height is the growth in mass of the incipient particles created at about the 1650 K isotherm. When the fuel is diluted with an inert, the flame temperature drops; and since the situation represents a moving-boundary (quasi-steady) heat-transfer problem, the 1650 K isotherm moves closer to the flame temperature isotherm. As a result, particle growth diminishes and the flame front closes. That is, because the mass of soot formed is smaller, it is consumed in the flame front, so no smoke height is observed. However, if the fuel mass is increased, more incipient particles form and, although growth is slower under dilute conditions, the soot mass again becomes large enough to exhibit a smoke height. By repeating the dilution procedure to further decrease the flame temperature, as stated, one can eliminate the smoke penetration, which can be induced again by adding more fuel.
Consider, for example, the fact that a coannular diffusion flame with acetylene will have a much higher flame temperature than that of butene. When considered at the centerline, the incipient particle temperature isotherm for acetylene is much farther from the flame temperature isotherm at the same position than is that of butene; that is, the acetylene particles formed have a larger growth path. Thus, although more incipient particles may form at about 1650 K for butene, the overall soot volume fraction from acetylene may be greater owing to a longer growth period. The depth of the thermal wave (or isotherm) is of the order α/u, where α is the thermal diffusivity and u is the linear velocity. There are self-compensating terms in each of these parameters even when there is dilution. Recall that the flame height for a diluted fuel varies to the first order solely with the volumetric flow rate of the fuel, even under buoyancy conditions. Figure 8.16 shows some results of this soot elimination procedure in which diluents other than nitrogen were chosen. Argon requires greater dilution to achieve the same temperature as nitrogen, and carbon dioxide requires less. Yet the variation in the incipient particle temperature for the four fuels shown in Figure 8.16 is slight and well within experimental error.
Because different fuels produce different amounts of incipient soot particles at about 1650 K, the smoke height test becomes a measure of the fuel's relative propensity to soot under flame-like conditions. However, if the smoke height is to be used for fuel comparisons, the diffusion flame temperature must be adjusted to be the same for all fuels. Further, based on the results of Harris and Weiner [79,80] for premixed flames, the assumption is made that the growth rate of particles is similar for all fuels that produce particles. Opposed-jet systems also permit good estimates of particle growth times along the jet stagnation streamline, as shown by the results of Vandsburger et al. [67,68], which revealed, as expected, that increasing the temperature (increasing the oxygen mole fraction in the opposed stream, i.e., the oxygen index) increases the soot yield and that ethene produces more soot than propane. The same variational trend in soot volume fraction with time was obtained with Wolfhard–Parker burners [64] as in premixed flames [79,80]. Particle number density trends are also the same for various burner systems. For similar fuels, the general agreement between coflow and counterflow results for soot volume fractions, number densities, and particle size ranges is reasonably good. Since residence time is reduced near the flame in counterflow burners in comparison with coflow systems, the maximum soot formation rates in counterflow systems are lower than those in coflow systems for comparable fuels and flame temperatures. When soot growth rates are normalized by the available soot surface area to give what Harris and Weiner [79,80] called the “specific surface growth rate,” the results of coflow burners agree not only with those of counter flow burners, but also with those of rich premixed flames [55].
Detailed optical measurements of the growth patterns both in coflow and counterflow diffusion flame configurations have provided great insight into the extent of particle growth after soot nucleation. Many research efforts in these configurations have been performed to evaluate how additives to the fuel jet would affect soot formation and growth by causing either flame temperature variations or chemical inhibition. Considering that most of these efforts used optical diagnostics, owing to the work of Axelbaum and Law [84], it was shown that these types of additive measurements had a greater effect on soot volume fraction developed than flame temperature control; that is, the additives affected the measurement rather than any soot process. This concept was verified as long as the resulting flame temperature was above the nucleation temperature. Of course, as discussed earlier, for diffusion flames, when the flame temperature is below the nucleation temperature (∼1650 K at 1 atm), the kinetic time scales for soot nucleation are too slow for particulate formation. In the context stated earlier, what controls the soot volume fraction that penetrates the laminar diffusion flame to cause the smoke height is the distance between the isotherms that specify the incipient particle formation (nucleation) and the stoichiometric flame temperature. Again, this distance establishes the growth time of the particles formed before flame oxidation of the soot occurs, or in the case of additives, allows soot volume fraction measurements to evaluate the possible effect of these additives on soot mass fraction trends. As stated, for the given gaseous flow rate in diffusion flames, a quasi-steady heat transfer condition exists that determines the temperature field within the burning, soot-forming jet. Consequently, the thermal diffusivity of the flowing gas mixture and its velocity are of direct importance to the overall soot volume fraction formed within the flame structure. Now considering conditions along the diffusion flame centerline streamline, a simple one-dimensional moving boundary problem exists and the temperature variation along the centerline (streamline) can be readily shown [85] to be
ln[(Tf−To)/(Tn−To)]=(u/α)z
(8.147)
where Tf, Tn, and To are the flame, incipient particle formation, and initial fuel jet temperatures, respectfully; u is the velocity along the axial streamline; z is the distance between Tf and Tn or the particle growth distance within the diffusion flame; and (α/u) is generally referred to as the depth of the thermal wave (field). The centerline is, of course, the most appropriate and simplest streamline to analyze. In a conical diffusion flame all other streamlines curve. Streamlines emanating near the wall of the fuel jet will curve to the extent that Tf and Tn essentially merge so there is an area of blue luminosity (no soot).
What is important to realize from Eqn (8.147) is that the flame temperature effect is in the log term, and thus variations in flame temperature are not significant with regard to increases of soot mass in diffusion flames. The extent of the thermal field is important; that is, the depth of the thermal wave (α/u) inside the burning fuel jet. Although in turbulent jet flames the flame isotherm is fluctuating and thus is not spatially fixed, but within a band, lifted or not, the general trend given by Eqn (8.147) has been found to be appropriate by experiments conducted [86]. In essence, then, physical conditions that affect fuel stream velocity and thermal diffusivity and their dependency on pressure and jet exit temperature dominate the extent of soot production in diffusion flame situations. Most early diffusion flame experimental work with respect to soot tendency was under small Froude number consideration, but has given insight into the laminar flame sooting process. Also, under the buoyancy condition the jet fuel exit velocity increases as the actual flame height is approached. Sooting flame experiments under microgravity conditions, which in essence is a completely momentum-controlled situation based on fuel exit velocity, show a bellowing flame structure as described in Figure 6.9, which is a result of the Burke–Schumann development. It follows that jet flames will soot more and appear wider in microgravity, as has been found [87]. As well, opposed jet diffusion flame work gives soot particle growth in an inverted temperature field compared with that given by Eqn (8.147). Consequently, the growth patterns in opposed jet diffusion flames, and for that matter in inverted coflow annular flames, will not emulate fuel jet injection soot characteristics.
The data of Axelbaum and Law [84] clearly support the concept of the thermal field effect and the effect of inert dilution on soot volume fraction measurements as developed by Eqn (8.147). Shown in Figure 8.17 are their soot volume fraction data for a 100% ethene jet (A), a 50% ethene–50% nitrogen jet (B), and a 50% ethane–50% nitrogen jet whose air was adjusted (C) to give the same temperature as (A). Notice the close correspondence between (B) and (C) even though the flame temperatures are vastly different. The effect of thermal diffusivity is best shown in some early soot measurements [88] as given by Figure 8.18. This plot correlates the ethene flow rate to reach the smoke height as a function of the added diluent flow rate. A lower ethene flow rate indicates a greater tendency to soot. At a fixed diluent flow rate that indicates a fixed average jet velocity, the results of Ar and N2 are practically the same even though the flame temperature would be appreciably higher for Ar. Interestingly, the thermal diffusivity of argon and nitrogen is practically the same. Note the difference in the Ar and He results where the temperatures are the same but the thermal diffusivity of He is an order of magnitude greater than Ar. The same aspect explains the difference between SO2 and CO2 in which the CO2 has a thermal diffusivity about a factor of two larger than SO2. Indeed, the concept exemplified by Eqn (8.147) explains much of the additive results in the literature. Remarkably, the simple smoke height experiments reveal the same trends without the necessity of the more complex soot volume fraction measurements. Of course, experiments to control the diffusion flame temperature by addition of inerts such as helium to the air stream will produce results different in experiments when the inert is added to the fuel stream. The reasoning is that addition to the fuel stream affects the overall thermal diffusivity in the thermal field within the fuel stream. For example, if the nitrogen from the air stream is transferred to the fuel stream, the same stoichiometric temperature would prevail in a fuel jet experiment. However, the additional volumetric flow of nitrogen will increase the fuel stream flow and decrease the depth of the thermal field (α/u). Thus, the extent of soot growth is reduced. The high oxygen content of the outer flow does not affect the flame structure chemistry, as some have claimed, and thus the soot mass development.
Figure 8.17Effect of dilution and temperature reduction, resulting from inert addition to the fuel, on soot formation. A: 100% Ethene, B: 50% Ethene / 50% Nitrogen, C: same as B except temperature has been adjusted to be equal that of A by replacing a portion of the nitrogen in the oxidizer with an equal molar amount of argon. From Ref. [84]
Exhaust gas recirculation in various furnaces and diesel operations has been used to control NOx and soot formation. With regard to soot formation the question arises as to the effect of induced oxygen or oxygen-containing species on the soot forming process. It is known that even in simple laminar fuel jets there are some slight amounts of oxygen infused into the fuel jet stream. It has been shown in fuel jet experiments that small amounts of oxygen only have an effect on tightly bound simple fuels such as ethene or ethyne, otherwise oxygen acts as a diluent until appreciable amounts enter the fuel stream [69]. The data shown in Figure 8.19 use peak extinction coefficients as a soot measurement for the cylindrical opposed jet diffusion flames. The oxygen index (i.e., the oxygen mole fraction in the oxidizer stream) is that which sets the adiabatic flame temperature of the diffusion flame for all oxygen additions on the fuel side. Notice for ethene that both for an oxygen index of 0.21 (air equivalent) and richer (0.18) that the addition of oxygen on the fuel side increases product formation as it accelerates ethene pyrolysis and oxidation and heat release. Then the mixture becomes essentially premixed on the fuel side, and as the fuel side temperature increases, the soot production decreases. For propane, however, addition of oxygen on the fuel side acts as a diluent until there is enough oxygen to induce chemical energy release. The oxygen index for the propane results was taken as 0.25 because propane does not have a smoke height in air. Smoke height results for ethene, propane, and isobutene correlate well with opposed jet results [69]. Most practical fuels have many constituents, which would indicate that some components would follow the trend indicated in Figure 8.19. Indeed, propane could be a simple surrogate fuel for more complex hydrocarbons in these type experiments. Thus, the analysis indicated in Eqn (8.147) would to a large extent correctly correlate results in high-pressure diesel-like chambers, as given in Ref. [90].
Figure 8.18The effect of inert diluents on sooting volume fraction. From Schug et al. [88].
In direct fuel injection diesel engine experiments the flame front lifts from the jet outlet, and one must realize that this condition does not affect the soot analysis as given by Eqn (8.147) since Tn and Tf lie well within the lifted flame structure. Indeed, application of the principle of Eqn (8.147) to turbulent lifted diesel flame-type experiments would explain many results reported in Ref. [83].
figure 8.19Peak extinction coefficient versus equivalence ratio of fuel/oxygen stream mixture in propane and ethene opposed-jet diffusion flames. fw is the fuel ejection parameter, which is a measure of mass flux from the burner. The greater the extinction coefficient, the greater the soot mass. From Ref. [89].
8.5.5. Detailed Structure of Sooting Flames
The critical sooting equivalence ratios for premixed flames and the smoke heights for diffusion flames discussed so far can serve only as qualitative measurements that offer excellent insights into the controlling factors in the soot incipient formation process. To gain greater understanding of the soot processes in flames, it has been necessary to undertake extensive chemical and physical measurements of temperature and velocity as well as laser extinction, scattering, and fluorescence in premixed flames [91,92], Wolfhard−Parker burners [93–95], coannular burners [96–98], and opposed-jet diffusion flames [67,68,89,99] (see Ref. [55]).
Harris and Weiner [79,80] contributed much to the study of soot particle history in premixed flames. They used laser light scattering and extinction measurements to determine particle size, number density, and volume fraction in experiments with flat flame burners stabilized by a water-cooled porous plug. Their studies showed that richer ethene flames produce more soot owing to a higher nucleation rate, increased surface growth in these richer flames is induced by the increased surface area available for growth, and depletion of growth species does not occur in these flames. Therefore, these investigators concluded that the final particle size reached when surface growth has virtually stopped is determined not by growth species depletion, but rather by a decrease in the reactivity of the particles. They further concluded that the results are applicable to other fuels because the post-flame gases of aliphatic fuels are all similar, acetylene and methane being the prominent hydrocarbons present. Thus, the similarity in surface growth in aliphatic fuels is related to the dominant role that acetylene plays in these processes. Although the post-flame gases of aromatic fuels have about 100 times more PAH than the post-flame gases of aliphatic fuels [100], acetylene remains the principal hydrocarbon in the burned gases of benzene flames [101]. This finding, combined with the fact that growth rates in benzene and ethylene are nearly identical [93], suggests that the mechanism of growth in benzene flames is also dominated by acetylene. These conclusions lend great validity to the qualitative results, postulates, and correlation given previously for premixed flames [57,102]. As stated, this surface growth pattern has been found to be the same in diffusion flames as well.
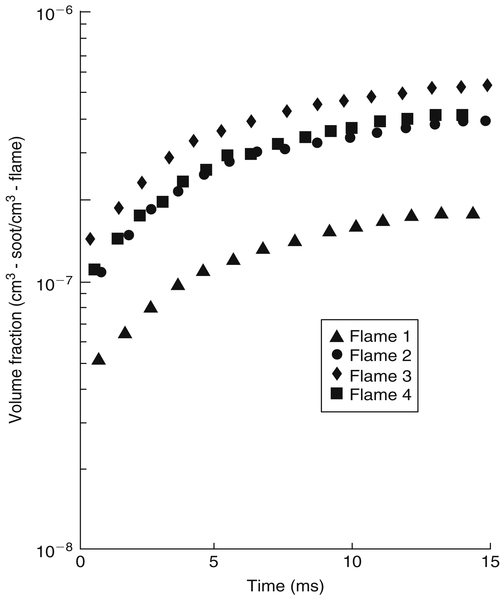
For the flames described in Table 8.7, Harris and Weiner [79] obtained the results shown in Figure 8.20, where the increase of soot volume fraction is plotted as a function of time. Thus, temperature measurements revealed that Flame 3 has the lowest temperature, Flames 2 and 4 are of equal and somewhat higher temperature, and Flame 1 has the highest temperature. These results supply quantitative proof that in premixed flames the tendency to soot decreases with increasing flame temperature. Also of importance is that Flame 2, which has toluene as a fuel constituent and the same equivalence ratio as that of Flame 4 for pure ethene, gives the same soot volume fraction as Flame 4 (Figure 8.20). A larger initial soot volume fraction indicates a larger initial incipient particle formation. The particle number density decreases with time owing to coagulation; however, fuels that have a larger initial number density (incipient particle formation) have the largest final number density. Similar results for particle histories were found in opposed-jet diffusion flames [67,68]. These quantitative results support the use of the qualitative critical sooting equivalence ratio results obtained on Bunsen flames as an excellent means for evaluating the sooting tendency of fuels under premixed conditions.
Figure 8.20Soot volume fraction profiles for premixed flames. See Table 8.7. From Harris and Weiner [79].
One of the earliest detailed diagnostic efforts on sooting of diffusion flames was that of Wagner et al. [93–95], who made laser scattering and extinction measurements, profile determinations of velocity by laser doppler velocimetry (LDV), and temperature measurements by thermocouples on a Wolfhard−Parker burner using ethene as the fuel. Their results showed clearly that soot particles are generated near the reaction zone and are convected farther toward the center of the fuel stream as they travel up the flame. The particle number densities and generation rates decline with distance from the flame zone. The soot formation rate appeared to peak at values about 2–3 mm on the fuel side of the reaction zone. The particle number density decreases by coagulation and gradually levels off. This process leads to an increase in particle size and a simultaneous increase in soot volume fraction as the particles absorb hydrocarbon species. Thus, the process is similar to that found by Harris and Weiner in premixed flames. Smyth et al. [64] drew essentially the same conclusions in characterizing a sooting methane Wolfhard−Parker burner by detailed species profiles.
Santoro et al. [96,97] performed some early extensive quantitative studies of coannular diffusion flames. They found that a striking feature of the coannular laminar diffusion flame is the existence of a roughly toroidal zone near the base of the flame where intense nucleation coexists with intense agglomeration to produce moderate-size particles. The soot formed in this region is convected upward along streamlines to form an annular region in which further gas-to-particle conversion occurs at moderate rates. Indeed, it is because of the intense particle formation in this toroidal region that the smoke height is identified as that point at which wings appear in the laminar diffusion flame [74]. The results of Santoro et al. [97], depicted in Figure 8.21, also indicate that the major soot formation regime is 2–3 mm on the fuel side of the flame, whose position is specified by the maximum temperature. In support of the concept postulated previously, one notes that the point of maximum particle density occurs at about 1600 K. That particles exist at other temperatures along the analysis profile in Figure 8.21 is attributable to thermophoresis.
Furthermore, fluorescence measurements aimed at detecting PAHs showed that the maxima clearly precede the maxima of soot volume fraction. No apparent difference in fluorescence levels was observed in experiments with the butene and benzene fuels, in contrast with the findings of other investigators [93]; thus, PAHs apparently have the same role in diffusion flames for both aliphatic and aromatic fuels. In fact, PAHs may be involved in the soot nucleation stage (as has often been hypothesized and will be discussed next); such a finding, together with the relative constancy of the temperature at the soot onset, would suggest that even though the extent of fuel to soot conversion may vary significantly from fuel to fuel, a common soot formation mechanism exists for all fuels.
Kent and Wagner [103] made some interesting quantitative measurements of overall axial soot concentrations as well as soot, flame zone, and centerline temperatures of coannular flames that have great significance. These results for ethene at different flow rates are shown in Figure 8.22. The data indicate that flames emit smoke when the soot temperature in the oxidation zone falls below 1300 K. As the temperature of the soot decreases, the flow proceeds downstream because of cooling and radiation losses because of the soot mass formed interior to the flame zone. Thus, the flow rates indicated in Figure 8.22 for 3.4 and 5.2 mL/s would have flames below the smoke height, and the actual smoke height would exist for a volumetric flow rate between 5.2 and 6.1 mL/s. Establishing that smoke is emitted when burnout ceases as the soot temperature drops below 1300 K, regardless of other conditions, is a significant and practical result. Thus, if incipient particle formation occurs in the 1650 K range under flame conditions and particle burnout ceases below about 1300 K range, a soot formation window exists for combustion processes. If one wishes to prevent soot from emanating from a combustion system operating in normal ranges, the temperature must be kept below about 1650 K. If this procedure is not possible, the exhaust must be kept lean and above 1300 K. With this understanding, one can visualize the Catch-22 problem facing diesel engine operators. When diesel engines are operated lean to reduce the thermal NOx the manifold exhaust temperature becomes too low to burn the soot particles that will form; but when they are operated less lean to burn the soot, the NOx increases. It is also interesting to recall from Chapter 3 that CO oxidation does not become rapid until above 1100 K, and as will be discussed in Chapter 9, a primary oxidant of soot is OH. Thus, the observed 1300 K when burnout ceases could also imply competition in the flame for OH radicals between CO and soot and consequently local extinguishment of CO oxidation when smoke is emitted.
Figure 8.21Comparison of radial profiles for scattering cross-section Qvv and temperature (uncorrected) as a function of radial position for a coannular ethane diffusion flame. From Santoro et al. [96].
Figure 8.22Axial distributions of relative soot yield x; maximum, centerline, and soot temperatures for ethene at flow rates around soot point in coaxial diffusion flames. Flow rates (ml/s): ⋄ 3.4, ∇ 5.2, ○ 6.1, Δ 7.2, □ 10.9. From Kent and Wagner [103].
8.5.6. Chemical Mechanisms of Soot Formation
The quantitative and qualitative results presented in the previous sections confirm some original arguments that the chemistry of fuel pyrolysis, and hence fuel structure, plays an important and possibly dominant role in sooting diffusion flames. The identical chemical character of the soot formed from diffusion or premixed controlled systems seems to suggest a commonality in the chemical mechanisms for soot formation. Apparently, there is an underlying fuel-independent general mechanism that is modified only with respect to alternative routes to intermediates. These routes are simply affected by the combustion system temperature and the general characteristics of the initial fuel. Essentially, this concept implies that the relative propensity of one fuel to soot compared with another is primarily determined by differences in the initial rate of formation of the first and second ring structures; moreover, while the mechanisms controlling the growth of large condensed-ring aromatics, soot nucleation, soot growth, etc., remain essentially unchanged, the growth steps in large aromatic structures that lead to soot nucleation are significantly faster than the initial-ring formation [104]. Thus, the formation of the initial aromatic rings controls the rate of incipient soot formation. As is well established now, the incipient soot formation particle concentration determines the soot volume fraction: that is, the total amount of soot formed. In support of this general contention that initial-ring formation is the soot-controlling step are the results correlating the sooting tendencies of several fuels as measured by time-of-flight mass spectrometry coupled to a shock tube and chemical sampling in coannular normal and inverse diffusion flames [104]. These cases revealed the presence of allene, which was established kinetically to form benzene directly through a C3 route [105]. Miller and Melius [106] also formulated a chemical kinetic model of soot formation in which they argued that the first aromatic ring is most likely formed by reaction of two propynyl (C3H3) radicals; correlations [66] of experimental results on the propensity of C3 hydrocarbon fuels to soot appear to support this route.
To explain the underlying principles of molecular growth to soot formation, mechanisms related to ions [107], ring growth [108,109], polyacetylene chains [71], Diels–Alder reactions, and neutral radicals [110] have been proposed [111]. However, the experimental results discussed on sooting tendencies of fuels under various flame configurations can be explained well by a generalized mechanism based on soot precursor growth through a system dependent on neutral radicals.
The detailed modeling of soot formation in the shock tube pyrolysis of acetylene [112] and other fuels [113] provides the central basis for the fuel-independent general mechanisms suggested here. Also, a large body of work by Howard and co-workers [114,115] on premixed flames with regard to formation of aromatic species provides direct tests of the proposed mechanisms and are key to understanding and modeling soot formation.
The dominant route, as proposed by Frenklach et al. [112], is the focus of this development. Indeed, this mechanism (Figure 8.23) would occur mostly at high temperatures [112] and serves as the preferred route in premixed flames where the original fuel structure breaks down to acetylene, and soot is formed in the high-temperature post-flame zone. Shock tube results by Colket [116] strongly suggest that formation of benzene from acetylene pyrolysis is largely attributable to acetylene addition to the n-butadienyl radical (C4H5), not C4H3. Thus, one could conclude that in diffusion flames where soot begins forming about 1600 K, the chemical route is alternate route A in Figure 8.23. Colket’s results [116] for vinylacetylene in the same temperature regime indicate that the main route to benzene is through vinyl radical addition to the vinylacetylene, as specified in Figure 8.23 by alternate route B.
These three paths offer excellent insight into the sooting tendencies measured in premixed, coflowing, and counterflowing diffusion flames and shock tubes. The fastest and most abundant route to the formation of the appropriate intermediates for the growth of the first aromatic ring determines the incipient particle formation rate and the relative propensity of a fuel to soot. By examining this fuel propensity to soot, it becomes evident that C4 hydrocarbons have a greater propensity to soot because they most readily form the butadienyl radical [71]. This hypothesis certainly explains the early results of Schalla et al. [61] for the n-olefin series in which butene had the smallest smoke height. Temperature control was not necessary in Schalla’s tests with olefins because all olefins have the same C/H ratio and hence near identical flame temperature histories. Butene, then, soots more than acetylene because it creates the butadienyl radical more readily. Butadiene soots more than either butene or acetylene, again because it more readily forms the butadienyl radical.
Figure 8.23General mechanisms for soot formation.
That most alkylated benzenes show the same tendency to soot is also consistent with a mechanism that requires the presence of phenyl radicals, concentrations of acetylene that arise from the pyrolysis of the ring, and the formation of a fused-ring structure. As mentioned, acetylene is a major pyrolysis product of benzene and all alkylated aromatics. The observation that 1-methylnaphthalene is one of the most prolific sooting compounds is likely explained by the immediate presence of the naphthalene radical during pyrolysis (see Figure 8.23).
Sampling in inverse coannular diffusion flames [66] in which propene was the fuel has shown the presence of large quantities of allene. Schalla et al. [61] also showed that propene is second to butene as the most prolific sooter of the n-olefins. Indeed, this result is consistent with the data for propene and allene in Ref. [77]. Allene and its isomer methylacetylene exhibit what at first glance appears to be an unusually high tendency to soot. However, Wu and Kern [117] showed that both pyrolyze relatively rapidly to form benzene. This pyrolysis step is represented as alternate route C in Figure 8.23.
Detailed examination of the chemical reactions in the general mechanism in Figure 8.23 reveals the importance of the pyrolysis fragments of all the fuels considered, especially the significance of H atoms, vinyl radicals, and acetylene. In particular, it becomes apparent that H atom and acetylene concentrations are important because they regulate, by abstraction and addition, respectively, the rapidity with which the large soot precursors can grow. The presence of large concentrations of H atoms in fuel pyrolysis is due to the thermal decay of the alkyl radicals present. Since the unimolecular cleavage of these radicals is not fast until temperatures well above 1100 K are reached [118], it is possible that this kinetic step and H atom diffusion set the range of the critical temperature for incipient soot formation in diffusion flames. Since there is no radical diffusion in shock tube studies of soot formation, it is not surprising, as mentioned, that the temperature at which soot formation is first identified is somewhat higher than that for diffusion flames.
8.5.7. Influence of Physical and Chemical Parameters on Soot Formation
The evidence suggests that temperature is the major parameter governing the extent to which a given fuel will soot under a particular flame condition or combustion process. As emphasized earlier, increasing the temperature under premixed fuel−air conditions decreases soot production, whereas the opposite is true for diffusion flames. The main effect of varying pressure must be manifested through its effect on the system temperature. Fuel pyrolysis rates are a function of concentration, but the actual dependence on the fractional fuel conversion depends on the overall order of the pyrolysis process. Unfortunately, varying the pressure alters the structure of the diffusion flame and the diffusivity of the fuel. As discussed, a variation in the diffusivity can shorten the growth period of the incipient particles formed. The diffusivity varies inversely with the pressure.
Regrettably, the particular effect of an additive is difficult to assess. A chosen diluent cannot lower the flame temperature without altering the effective thermal diffusivity of the fuel−diluent jet. Thus, in examining the possible effect of an additive it is imperative to consider how the measurements of the extent of sooting were made. With respect to physical parameters, the effect of temperature is clear. However, the data reported on pressure variation must be viewed with caution since, as stated, pressure variations cause variations in temperature, thermal diffusivity, flow velocity, and flame structure.
It now seems evident that additives that alter the early concentrations of H atoms can have a recognizable effect on the soot output. Again, the extent of the effect depends on the type of flame, the specific fuel, and the addition. Perhaps the best example is the addition of modest amounts of oxygen to the fuel in a diffusion flame configuration. The addition of oxygen to tightly bound fuels such as ethene, ethyne, and benzene noticeably affects the sooting tendency of these fuels. Similarly, a modest amount of oxygen added to the paraffins tends to act as an inert diluent [69]. Chemical kinetic analyses of the pyrolysis rates of these fuels at modest temperatures confirm that the effect in the case of the tightly bound fuels is to rapidly increase the H atom concentration, whereas rapid creation of H atoms owing to their thermal homolysis of paraffins is little affected by the presence of oxygen. The general effect of continuously increasing the oxygen concentration on the fuel side of ethene and propane in opposed-jet diffusion flame experiments supports these conclusions and gives an overall picture of the effect of oxygen addition [69]. In these experiments, as the fuel−oxygen ratio was increased, the oxygen concentration in the opposed jet was reduced to keep the flame temperature constant. The particular results are shown in Figure 8.19. The higher the extinction coefficients, the greater are the soot volume fractions. Ethene’s propensity to soot rises with decreasing ϕ (small oxygen addition). As ϕ decreases further, a chemical reaction releases energy, altering the temperature profile and increasing the sooting tendency still further. At a given ϕ, oxidizer species become present in sufficient abundance that the soot precursors are attacked. Eventually, oxidizer attack exceeds the thermal effect, a maximum is reached, and the conditions approach those of a premixed flame. The soot formed then decreases with decreasing ϕ, vanishing at a value near the critical sooting equivalence ratio determined by the conventional methods discussed. The same trends hold for propane, except that the oxygen initially acts as a diluent, lowering soot production until the reaction is sufficiently initiated to raise the overall thermal profile level.
The results in Figure 8.19 are significant in that for values of ϕ less than the maximum shown, premixed flame conditions exist. Since the flammability limit lies between the critical sooting equivalence ratio and the maximum ϕ for sooting, it is possible to conclude that under all premixed flame conditions, fuel structure plays no role in the sooting tendency. Also, when ϕ decreases near infinity, the sooting tendency rises for ethene and decreases not only for propane, but would also for n-butane and isobutane [69]. The fact that these opposing trends can be explained by a neutral radical reaction mechanism tends to support the notion that such mechanisms control the soot formation rate. It is difficult to see how the trends could be explained on the basis of an ion mechanism. Furthermore, the wide variation of sooting tendency with ϕ indicates the difficulty of extracting fundamental information from studies of turbulent fuel jets.
The presence of halogen additives substantially increases the tendency of all fuels to soot under diffusion flame conditions [73]. The presence of H atoms increases the soot pyrolysis rate because the abstraction reaction of H + RH is much faster than R + RH, where R is a hydrocarbon radical. Halogenated compounds added to fuels generate halogen atoms (X) at modest temperatures. The important point is that X + RH abstraction is faster than H + RH, so that the halogen functions as a homogeneous catalyst through the system
X+RH→R+XH
H+XH→H2+X
Sulfur trioxide is known to suppress soot in diffusion flames and increase soot in premixed flames. These opposite effects can be explained in the context of the section on sulfur oxidation (Section 8.4.2). In diffusion flames, the slight suppression can be attributed to the reaction of H atoms via the step
H+SO3→OH+SO2
If this step occurs late in the pyrolysis process, the hydroxyl radicals that form could attack the soot precursors. Thermal diffusivity may also have an effect. In premixed flames the SO3 must dissociate into SO2, which removes H atoms by
H+SO2+M→HSO2+M
The reduction in H concentration leads to a decrease in OH, and hence to an increase in soot production.
As additives to reduce soot output in flames, metal and organometallic compounds such as ferrocene have attracted the attention of many investigators (see Ref. [119]). The effect in premixed flames is best described by Bonczyk [120], who reported that the efficiency with which a given metal affects soot production characteristics depends almost exclusively on temperature and the metal atom’s ionization potential. Thus the alkaline-earth metals Ba > Sr > Ca are the most effective. The metal additives reduce the soot particle size and volume fraction and increase the number density. Essentially, ion charge transfer reduces agglomeration, and under the appropriate conditions, particle burnup is augmented. Indeed, the burnup effect is prevalent in diffusion flames since the smaller particles must pass through the flame front. The conditions describing the smoke height are pertinent. There is no clear evidence that metal additives affect the incipient particle formation in diffusion flames. The difficulty in understanding the effect of ferrocene in diffusion flames is compounded by the fact that it decomposes into cyclopendienyl, which readily forms soot.