
3.8. The Oxidation of Higher-Order Hydrocarbons
3.8.1. Aliphatic Hydrocarbons
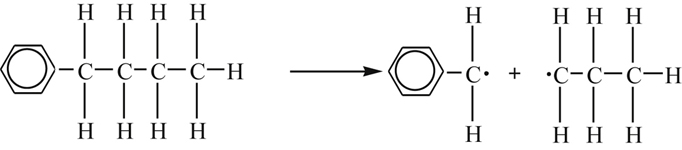
The high-temperature oxidation of paraffins larger than methane is a fairly complicated subject owing to the greater instability of the higher-order alkyl radicals and the great variety of minor species that can form (see Table 3.2). But, as is the case with methane [32–35], there now exist detailed models of ethane [40], propane [41], and many other higher-order aliphatic hydrocarbons (see for example Ref. [42]). Despite these complications, it is possible to develop a general framework of important steps that elucidate this complex subject.
3.8.1.1. Overall view
It is interesting to review a general pattern for oxidation of hydrocarbons in flames, as suggested very early by Fristrom and Westenberg [43]. They suggested two essential thermal zones: the primary zone, in which the initial hydrocarbons are attacked and reduced to products (CO, H2, H2O) and radicals (H, O, OH), and the secondary zone, in which CO and H2 are completely oxidized. The intermediates are said to form in the primary zone. Initially, then, hydrocarbons of lower order than the initial fuel appear to form in oxygen-rich, saturated-hydrocarbon flames according to
![]()
Because hydrocarbon radicals of higher order than ethyl are unstable, the initial radical CnH2n+l usually splits off CH3 and forms the next lower-order olefinic compound, as shown. With hydrocarbons of higher order than C3H8, there is fission into an olefinic compound and a lower-order radical. Alternatively, the radical splits off CH3. The formaldehyde that forms in the oxidation of the fuel and of the radicals is rapidly attacked in flames by O, H, and OH so that formaldehyde is usually found only as a trace in flames.
Fristrom and Westenberg claimed that the situation is more complex in fuel-rich saturated-hydrocarbon flames, although the initial reaction is simply the H abstraction analogous to the preceding OH reaction, for example,
![]()
Table 3.2
Relative Importance of Intermediates in Hydrocarbon Combustion
| Fuel | Relative Hydrocarbon Intermediate Concentrations |
| Ethane Propane Butane Hexane 2-Methylpentane | Ethene ≥ methane Ethene > propene ≥ methane > ethane Ethene > propene ≥ methane > ethane Ethene > propene > butene > methane ≥ pentene > ethane Propene > ethene > butene > methane ≥ pentene > ethane |
Under these conditions the concentrations of H and other radicals are large enough that their recombination becomes important, and hydrocarbons of order higher than the original fuel are formed as intermediates.
The general features suggested by Fristrom and Westenberg were confirmed [14,44] by high-temperature flow-reactor studies. However, this work permits more detailed understanding of the high-temperature oxidation mechanism and shows that under oxygen-rich conditions the initial attack by O atoms must be considered as well as the primary OH attack. More importantly, however, it has been established that the paraffin reactants produce intermediate products that are primarily olefinic, and the fuel is consumed, to a major extent, before significant energy release occurs. The higher the initial temperature, the greater the energy release, as the fuel is being converted. This observation leads one to conclude that the olefin oxidation rate simply increases more appreciably with temperature; that is, the olefins are being oxidized while they are being formed from the fuel. Typical flow-reactor data for the oxidation of ethane and propane are shown in Figures 3.12 and 3.13.
The evidence in Figures 3.12 and 3.13 [14,44] indicates three distinct, but coupled zones in hydrocarbon combustion:
1. Following ignition, the primary fuel disappears with little or no energy release and produces unsaturated hydrocarbons and hydrogen. A little of the hydrogen is concurrently oxidized to water.
2. Subsequently, the unsaturated compounds are further oxidized to carbon monoxide and hydrogen. Simultaneously, the hydrogen present and formed is oxidized to water.
3. Finally, the large amounts of carbon monoxide formed are oxidized to carbon dioxide and most of the heat released from the overall reaction is obtained. Recall that the CO is not oxidized to CO2 until most of the fuel is consumed owing to the rapidity with which OH reacts with the fuel compared to its reaction to CO (see Table 3.1).
3.8.1.2. Paraffin oxidation
In the high-temperature oxidation of large paraffin molecules, the chain initiation step is one in which a C–C bond is broken to form hydrocarbon radicals, namely,
![]() (3.98)
(3.98)
This step will undoubtedly dominate, since the C–C bond is substantially weaker than any of the C–H bonds in the molecule. As mentioned in the previous section, the radicals R′ and R″ (fragments of the original hydrocarbon molecule RH) decay into olefins and H atoms. At any reasonable combustion temperature, some C–H bonds are broken and H atoms appear owing to the initiation step
![]() (3.99)
(3.99)

For completeness, one could include a lower-temperature abstraction initiation step
![]() (3.100)
(3.100)
The essential point is that the initiation steps provide H atoms that react with the oxygen in the system to begin the chain branching propagating sequence that nourishes the radical reservoir of OH, O, and H; that is, the reaction sequences for the complete H2–O2 system must be included in any high-temperature hydrocarbon mechanism. Similarly, when CO forms, its reaction mechanism must be included as well.
Once the radical pool forms, the disappearance of the fuel is controlled by the reactions
![]() (3.101)
(3.101)
![]() (3.102)
(3.102)
where, again, X is any radical. For the high-temperature condition, X is primarily OH, O, H, and CH3. Since the RH under consideration is a multicarbon compound, the character of the radical R formed depends on which hydrogen in the molecule is abstracted. Furthermore, it is important to consider how the rate of reaction (3.102) varies as X varies, since the formation rates of the alkyl isomeric radicals may vary.
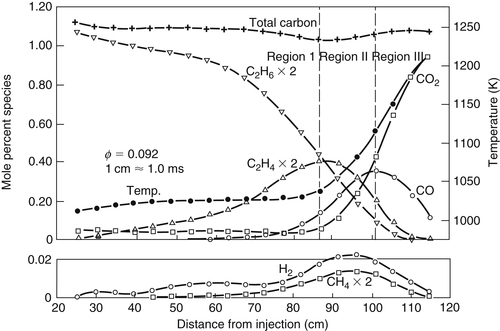
Data for the specific rate coefficients for abstraction from C–H bonds have been derived from experiments with hydrocarbons with different distributions of primary, secondary, and tertiary C–H bonds. A primary C–H bond is one on a carbon that is only connected to one other carbon, that is, the end carbon in a chain or a branch of a chain of carbon atoms. A secondary C–H bond is one on a carbon atom connected to two others, and a tertiary C–H bond is on a carbon atom that is connected to three others. In a chain, the C–H bond strength on the carbons second from the ends is a few kilojoules less than other secondary atoms. The tertiary C–H bond strength is still less, and the primary is the greatest. Assuming additivity of these rates, one can derive specific reaction rate constants for abstraction from the higher-order hydrocarbons by H, O, OH, and HO2 [45].
From the rates given in Ref. [45], the relative magnitudes of rate constants for abstraction of H by H, O, OH, and HO2 species from single tertiary, secondary, and primary C–H bonds at 1080 K have been determined [46]. These relative magnitudes, which should not vary substantially over modest ranges of temperatures, were found to be as listed here:
| Tertiary | : | Secondary | : | Primary | |
| H | 13 | : | 4 | : | 1 |
| O | 10 | : | 5 | : | 1 |
| OH | 4 | : | 3 | : | 1 |
| HO2 | 10 | : | 3 | : | 1 |
Note that the OH abstraction reaction, which is more exothermic than the others, is the least selective of H atom position in its attack on large hydrocarbon molecules. There is also great selectivity by H, O, and HO2 between tertiary and primary C–H bonds. Furthermore, estimates of rate constants at 1080 K [45] and radical concentrations for a reacting hydrocarbon system [47] reveal that the k values for H, O, and OH are practically the same and that during early reaction stages, when concentrations of fuel are large, the radical species concentrations are of the same order of magnitude. Only the HO2 rate constant departs from this pattern, being lower than the other three. Consequently, if one knows the structure of a paraffin hydrocarbon, one can make estimates of the proportions of various radicals that would form from a given fuel molecule (from the abstraction reaction (3.102)). The radicals then decay further according to
![]() (3.103)
(3.103)
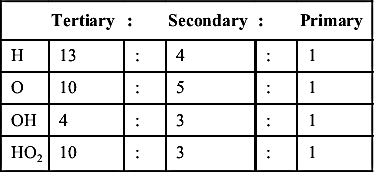
where R′ is an H atom or another hydrocarbon radical. The ethyl radical will thus become ethene and an H atom. Propane leads to an n-propyl and isopropyl radical:
These radicals decompose according to the β-scission rule, which implies that the bond that will break is one position removed from the radical site so that an olefin can form without a hydrogen shift. Thus the isopropyl radical gives propene and an H atom, while the n-propyl radical gives ethene and a methyl radical. The β-scission rule states that when there is a choice between a C–C single bond and a C–H bond, the C–C bond is normally the one that breaks because it is weaker than the C–H bond. Even though there are six primary C–H bonds in propane and these are somewhat more tightly bound than the two secondary ones, one finds substantially more ethene than propene as an intermediate in the oxidation process. The experimental results [14] shown in Figure 3.13 verify this conclusion. The same experimental effort found the olefin trends shown in Table 3.2. Note that it is possible to estimate the order reported from the principles just described.
If the initial intermediate or the original fuel is a large monoolefin, the radicals will abstract H from those carbon atoms that are singly bonded because the C–H bond strengths of doubly bonded carbons are large (see Appendix D). Thus, the evidence [14,46] is building that, during oxidation, all nonaromatic hydrocarbons primarily form ethene and propene (and some butene and isobutene) and that the oxidative attack that eventually leads to CO is almost solely from these small intermediates. Thus, the study of ethene oxidation is crucially important for all alkyl hydrocarbons.
It is also necessary to explain why there are parentheses around the collision partner M in reactions (3.98), (3.99) and (3.103). When RH in reactions (3.98) and (3.99) is ethane and R in reaction (3.103) is the ethyl radical, the reaction order depends on the temperature and pressure range. Reactions (3.98), (3.99) and (3.103) for the ethane system are in the falloff regime for most typical combustion conditions. Reactions (3.98) and (3.99) for propane may lie in the falloff regime for some combustion conditions; however, around 1 atm, butane and larger molecules pyrolyze near their high-pressure limits [48] and essentially follow first-order kinetics. Furthermore, for the formation of the olefin, an ethyl radical in reaction (3.103) must compete with the abstraction reaction
![]() (3.104)
(3.104)
Owing to the great instability of the radicals formed from propane and larger molecules, reaction (3.103) is fast and effectively first-order; thus, competitive reactions similar to reaction (3.100) need not be considered. Thus, in reactions (3.98) and (3.99) the M has to be included only for ethane and, to a small degree, propane, and in reaction (3.103) M is required only for ethane. Consequently, ethane is unique among all paraffin hydrocarbons in its combustion characteristics. For experimental purposes, then, ethane (like methane) should not be chosen as a typical hydrocarbon fuel.
3.8.1.3. Olefin and acetylene oxidation
Following the discussion from the preceding section, consideration will be given to the oxidation of ethene and propene (when a radical pool already exists) and, since acetylene is a product of this oxidation process, to acetylene as well. These small olefins and acetylene form in the oxidation of a paraffin or any large olefin. Thus, the detailed oxidation mechanisms for ethane, propane, and other paraffins necessarily include the oxidation steps for the olefins [42].
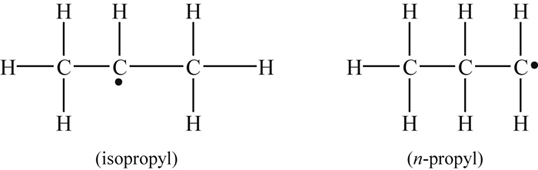
The primary attack on ethene is by addition of the biradical O, although abstraction by H and OH can play some small role. In adding to ethene, O forms an adduct [49] that fragments according to the scheme
The primary products are methyl and formyl radicals [50,51] because potential energy surface crossing leads to an H shift at combustion temperatures [49]. It is rather interesting that the decomposition of cyclic ethylene oxide proceeds through a route in which it isomerizes to acetaldehyde and readily dissociates into CH3 and HCO. Thus, two primary addition reactions that can be written are
![]() (3.105)
(3.105)
![]() (3.106)
(3.106)
Another reaction—the formation of an adduct with OH—has also been suggested [52]:
![]() (3.107)
(3.107)
However, this reaction has been questioned [4] because it is highly endothermic.
OH abstraction via
![]() (3.108)
(3.108)
could have a rate comparable to the preceding three addition reactions, and H abstraction
![]() (3.109)
(3.109)
could also play a minor role. Addition reactions generally have smaller activation energies than abstraction reactions; so at low temperatures the abstraction reaction is negligibly slow, but at high temperatures the abstraction reaction can dominate. Hence, the temperature dependence of the net rate of disappearance of reactants can be quite complex.
The vinyl radical (C2H3) decays to acetylene primarily by
![]() (3.110)
(3.110)
but, again, under particular conditions the abstraction reaction
![]() (3.111)
(3.111)
must be included. Other minor steps are given in Appendix C.
Since the oxidation mechanisms of CH3, H2CO (formaldehyde), and CO have been discussed, only the fate of C2H2 and CH2 (methylene) remains to be determined.
The most important means of consuming acetylene for lean, stoichiometric, and even slightly rich conditions is again by reaction with the biradical O [51,53,54] to form a methylene radical and CO
![]() (3.112)
(3.112)
through an adduct arrangement as described for ethene oxidation. The rate constant for reaction (3.112) would not be considered large in comparison with that for reaction of O with either an olefin or a paraffin. Mechanistically, reaction (3.112) is of significance. Since the C2H2 reaction with H atoms is slower than H + O2, the oxidation of acetylene does not significantly inhibit the radical pool formation. Also, since its rate with OH is comparable to that of CO with OH, C2H2—unlike the other fuels discussed—will not inhibit CO oxidation. Therefore, substantial amounts of C2H2 can be found in the high-temperature regimes of flames. Reaction (3.112) states that acetylene consumption depends on events that control the O atom concentration. As discussed in Chapter 8, this fact has implications for acetylene as the soot-growth species in premixed flames. Acetylene–air flame speeds and detonation velocities are fast primarily because high temperatures evolve, not necessarily because acetylene reaction mechanisms contain steps with favorable rate constants. The primary candidate to oxidize the methylene formed is O2 via
![]() (3.113)
(3.113)
however, some uncertainty attaches to the products as specified.
Numerous other possible reactions can be included in a very complete mechanism of any of the oxidation schemes of any of the hydrocarbons discussed. Indeed, the very fact that hydrocarbon radicals form is evidence that higher-order hydrocarbon species can develop during an oxidation process. All these reactions play a very minor, albeit occasionally interesting, role; however, their inclusion here would detract from the major steps and important insights necessary for understanding the process.
With respect to propene, it has been suggested [49] that O atom addition is the dominant decay route through an intermediate complex in the following manner:
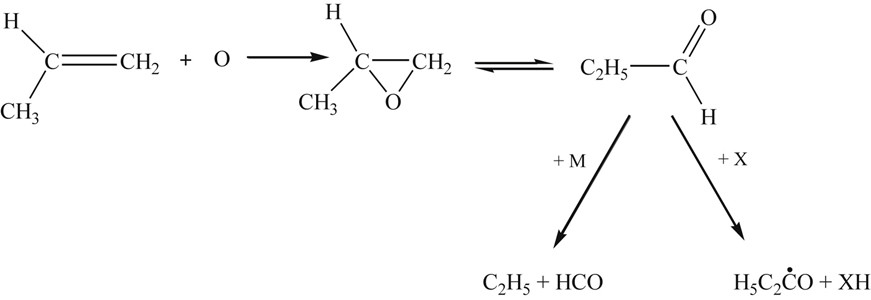
For the large activated propionaldehyde molecule, the pyrolysis step appears to be favored and the equilibrium with the propylene oxide shifts in its direction. The products given for this scheme appear to be consistent with experimental results [52]. The further reaction history of the products has already been discussed.
Essentially, the oxidation chemistry of the aliphatics higher than C2 has already been discussed, since the initiation step is mainly CC bond cleavage with some CH bond cleavage. But the initiation steps for pure ethene or acetylene oxidation are somewhat different. For ethene, the major initiation steps are [4,54]
![]() (3.114)
(3.114)
![]() (3.115)
(3.115)
Reaction (3.114) is the fastest, but reaction (3.115) would start the chain. Similarly, the acetylene initiation steps [4] are
![]() (3.116)
(3.116)
![]() (3.117)
(3.117)
Reaction (3.116) dominates under dilute conditions and reaction (3.117) is more important at high fuel concentrations [4].
The subsequent history of C2H and C4H3 is not important for the oxidation scheme once the chain system develops. Nevertheless, the oxidation of C2H could lead to chemiluminescent reactions that form CH and C2, the species responsible for the blue-green appearance of hydrocarbon flames. These species may be formed by the following steps [55–57]:
![]()
![]()
![]()
![]()
![]()
where the asterisk (∗) represents electronically excited species.
Taking all these considerations into account, it is possible to postulate a general mechanism for the oxidation of aliphatic hydrocarbons, namely,
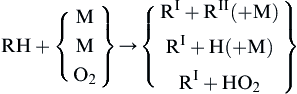
where the H creates the radical pool (X = H, O, and OH) and the following occurrences:
![]()
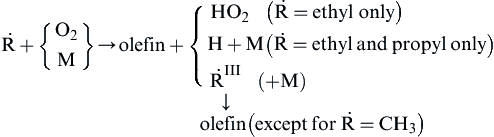
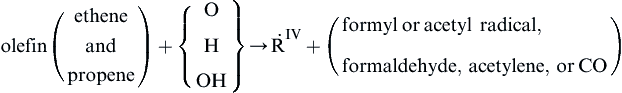
![]()
![]()
As a matter of interest, the oxidation of the diolefin butadiene appears to occur through O atom addition to a double bond as well as through abstraction reactions involving OH and H. Oxygen addition leads to 3-butenal and finally alkyl radicals and CO. The alkyl radical is oxidized by O atoms through acrolein (propenal) to form CO, acetylene, and ethene. The abstraction reactions lead to a butadienyl radical and then vinylacetylene. The butadienyl radical is now thought to be important in aromatic ring formation processes in soot generation [58–60]. Details of butadiene oxidation are presented in Ref. [61].
3.8.2. Alcohols
The presence of the OH group in alcohols makes alcohol combustion chemistry an interesting variation of the analogous paraffin hydrocarbon. The oxygen atom in the molecular structure generally alters the electronic structure, and consequently many of the C–H bond strengths are different than their values for structurally similar nonoxygenated hydrocarbons. Two fundamental pathways can exist in the initial attack on alcohols. In one, the OH group can be displaced while an alkyl radical also remains as a product. In the other, the alcohol is attacked at a different site and forms an intermediate oxygenated species, typically an aldehyde. The dominant pathway depends on the bond strengths in the particular alcohol molecule and on the overall stoichiometry that determines the relative abundance of the reactive radicals.
For methanol, the alternative initiating mechanisms are well established [67–71]. The dominant initiation step is the high-activation process
![]() (3.118)
(3.118)
which contributes little to the products in the intermediate (∼1000 K) temperature range [69]. By means of deuterium labeling, Aders [72] has demonstrated the occurrence of OH displacement by H atoms:
![]() (3.119)
(3.119)
This reaction may account for as much as 20% of the methanol disappearance under fuel-rich conditions [69]. The chain branching system originates from the reactions
![]()
which together are sufficient, with reaction (3.121) below, to provide the chain. As in many hydrocarbon processes, the major oxidation route is by radical abstraction. In the case of methanol, this yields the hydroxymethyl radical and, ultimately, formaldehyde via
![]() (3.120)
(3.120)
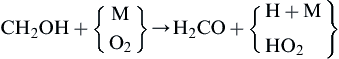 (3.121)
(3.121)
where as before X represents the radicals in the system. Radical attack on CH2OH is slow because the concentrations of both radicals are small owing to the rapid rate of reaction (3.120). Reactions of OH and H with CH3OH to form CH3O (vs CH2OH), and H2O and H2, respectively, have also been found to contribute to the consumption of methanol [70]. These radical steps are given in Appendix C.
The mechanism of ethanol oxidation, while not as well understood as methanol oxidation, has now been studied in flow and stirred reactors, shock tubes, and flames over a wide range of temperature and pressure. Although in flow reactor studies [62] acetaldehyde appears earlier in the reaction than does ethene, both species are assumed to form directly from ethanol. Studies of acetaldehyde oxidation [73] do not indicate any direct mechanism for the formation of ethene from acetaldehyde.
Because C–C bonds are weaker than the C–OH bond, ethanol, unlike methanol, does not lose the OH group in an initiation step. The dominant initial step leading to formation of the radical pool is
![]() (3.122)
(3.122)
As in all long-chain fuel processes, this initiation step does not appear to contribute significantly to the product distribution and, indeed, no formaldehyde is observed experimentally as a reaction intermediate. There is also indication in flames [65] that H2O elimination through a complex fission reaction involving a four-centered transition state can yield ethene directly
![]()
Hydrogen abstraction can lead to formation of three isomers of C2H5O. The favored reaction is abstraction of an H atom from the α C–H bond due to the weakness of this bond strength (∼397 kJ/mol) in the proximity of the electrophilic O atom. The CH3CHOH radical is then consumed by dissociation or reaction with O2.
![]() (3.123)
(3.123)
 (3.124)
(3.124)
The β C–H bond is the next weakest (∼424 kJ/mol), and because of the greater number of H atoms linked to the β carbon, abstraction of hydrogen at these sites yields CH2CH2OH, which thermally decomposes to ethene.
![]() (3.125)
(3.125)
![]() (3.126)
(3.126)
While the O–H bond energies (∼436 kJ/mol) are greater than C–H bond energies, hydrogen abstraction from the OH group produces the ethoxy radical (CH3CH2O), which along with reaction (3.124), produces acetaldehyde via β-scission of one C–H bond.
![]() (3.127)
(3.127)
![]() (3.128)
(3.128)
As described earlier, the acetaldehyde is consumed by either thermal decomposition or H atom abstraction
![]() (3.129)
(3.129)
![]() (3.130)
(3.130)
with the acetyl and CH2CHO radicals thermally dissociating.
Because the initial oxygen concentration determines the relative abundance of specific abstracting radicals, ethanol oxidation, like methanol oxidation, shows a variation in the relative concentration of intermediate species according to the overall stoichiometry. The ratio of acetaldehyde to ethene increases for lean mixtures.
As the chain length of the primary alcohols increases, thermal decomposition through fracture of C–C bonds becomes more prevalent. In the pyrolysis of n-butanol, following the rupture of the C3H7–CH2OH bond, the species found are primarily formaldehyde and small hydrocarbons. However, because of the relative weakness of the C–OH bond at a tertiary site, t-butyl alcohol loses its OH group quite readily. In fact, the reaction
![]() (3.131)
(3.131)
serves as a classic example of unimolecular thermal decomposition.
In the oxidation of t-butanol, acetone and isobutene appear [63] as intermediate species. Acetone can arise from two possible sequences. In one,
![]() (3.132)
(3.132)
![]() (3.133)
(3.133)
![]() (3.134)
(3.134)
![]() (3.135)
(3.135)
3.8.3. Aromatic Hydrocarbons
As discussed by Brezinsky [83], the oxidation of benzene and alkylated aromatics poses a problem different from the oxidation of aliphatic fuels. The aromatic ring provides a site for electrophilic addition reactions that effectively compete with the abstraction of H from the ring itself or from the side chain. When the abstraction reactions involve the side chain, the aromatic ring can strongly influence the degree of selectivity of attack on the side-chain hydrogens. At high enough temperatures, the aromatic ring thermally decomposes and thereby changes the whole nature of the set of hydrocarbon species to be oxidized. As will be discussed in Chapter 4, in flames the attack on the fuel begins at temperatures below those where pyrolysis of the ring would be significant. As the following sections will show, the oxidation of benzene can follow a significantly different path than that of toluene and other higher alkylated aromatics. In the case of toluene, its oxidation bears a resemblance to that of methane; thus it, too, is different from benzene and other alkylated aromatics.
In order to establish certain terms used in defining aromatic reactions, consider the following, where the structure of benzene is represented by the symbol  .
.
Abstraction reaction:
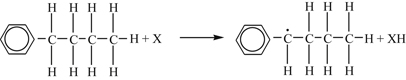
Displacement reaction:
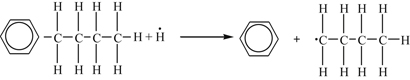
Homolysis reaction:
Addition reaction:

3.8.3.1. Benzene oxidation
Based on the early work of Norris and Taylor [84], and Bernard and lbberson [85], who confirmed the theory of multiple hydroxylation, a general low-temperature oxidation scheme was proposed [86,87], namely,
![]() (3.136)
(3.136)
![]() (3.137)
(3.137)
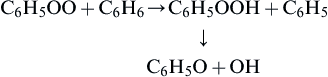 (3.138)
(3.138)
![]() (3.139)
(3.139)
![]() (3.140)
(3.140)
There are two dihydroxy benzenes that can result from reaction (3.140)—hydroquinone and pyrocatechol. It has been suggested that they react with oxygen in the following manner [84]:
 (3.141)
(3.141)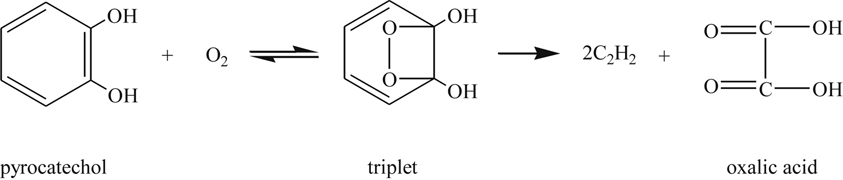 (3.142)
(3.142)Thus maleic acid forms from the hydroquinone and oxalic acid forms from pyrocatechol. However, the intermediate compounds are triplets, so the intermediate steps are “spin-resistant” and may not proceed in the manner indicated. The intermediate maleic acid and oxalic acid are experimentally detected in this low-temperature oxidation process. Although many of the intermediates were detected in low-temperature oxidation studies, Benson [88] determined that the ceiling temperature for bridging peroxide molecules formed from aromatics was of the order of 300 °C; that is, the reverse of reaction (3.137) was favored at higher temperatures.
It is interesting to note that maleic acid dissociates to two carboxyl radicals and acetylene
![]() (3.143)
(3.143)
while oxalic acid dissociates into two carboxyl radicals
![]() (3.144)
(3.144)
Under this low-temperature condition, the carboxylic radical undergoes attack
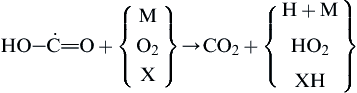 (3.145)
(3.145)
to produce CO2 directly rather than through the route of CO oxidation by OH characteristic of the high-temperature oxidation of hydrocarbons.
High-temperature flow-reactor studies [89,90] on benzene oxidation revealed a sequence of intermediates, which followed the following order: phenol, cyclopentadiene, vinylacetylene, butadiene, ethene, and acetylene. Since the sampling techniques used in these experiments could not distinguish unstable species, the intermediates could have been radicals that reacted to form a stable compound, most likely by hydrogen addition in the sampling probe. The relative time order of the maximum concentrations, while not the only criterion for establishing a mechanism, has been helpful in the modeling of many oxidation systems [4,15].
As stated earlier, the benzene molecule is stabilized by strong resonance; consequently, removal of an H from the ring by pyrolysis or O2 abstraction is difficult and hence slow. It is not surprising, then, that the induction period for benzene oxidation is longer than that for alkylated aromatics. The high-temperature initiation step is similar to that of all the cases described before, that is,
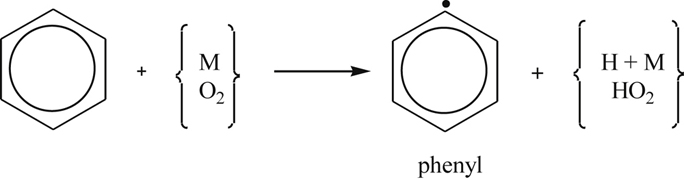 (3.146)
(3.146)but it probably plays a small role once the radical pool builds from the H obtained. Subsequent formation of the phenyl radical arises from the propagating step
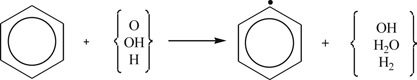 (3.147)
(3.147)The O atom could venture through a displacement and possibly an addition [89] reaction to form a phenoxy radical and phenol according to the steps
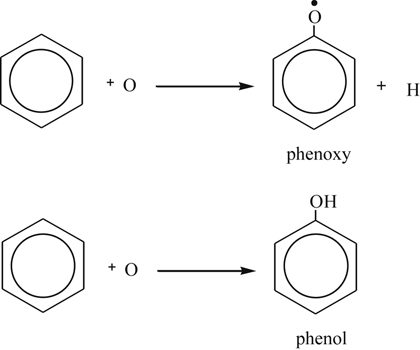 (3.148)
(3.148)Phenyl radical reactions with O2, O, or HO2 seem to be the most likely candidates for the first steps in the aromatic ring-breaking sequence [83,90]. A surprising metathesis reaction that is driven by the resonance stability of the phenoxy product has been suggested from flow reactor studies [83] as a key step in the oxidation of the phenyl radical:
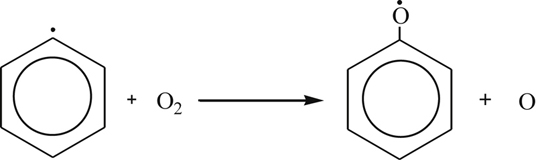 (3.149)
(3.149)In comparison, the analogous reaction written for the methyl radical is highly endothermic.
This chain branching step was found [90,91] to be exothermic to the extent of approximately 46 kJ/mol, to have a low activation energy and to be relatively fast. Correspondingly, the main chain branching step (reaction (3.21)) in the H2–O2 system is endothermic to about 65 kJ/mol. This rapid reaction (3.147) would appear to explain the large amount of phenol found in flow reactor studies. In studies [92] of near-sooting benzene flames, the low mole fraction of phenyl found could have required an unreasonably high rate of reaction (3.147). The difference could be due to the higher temperatures, and hence the large O atom concentrations, in the flame studies.
The cyclopentadienyl radical could form from the phenoxy radical by
 (3.150)
(3.150)The expulsion of CO from ketocyclohexadienyl radical is also reasonable, not only in view of the data of flow reactor results but also in view of other pyrolysis studies [93]. The expulsion indicates the early formation of CO in aromatic oxidation, whereas in aliphatic oxidation CO does not form until later in the reaction after the small olefins form (see Figures 3.12 and 3.13). Since resonance makes the cyclopentadienyl radical very stable, its reaction with an O2 molecule has a large endothermicity. One feasible step is reaction with O atoms, namely,
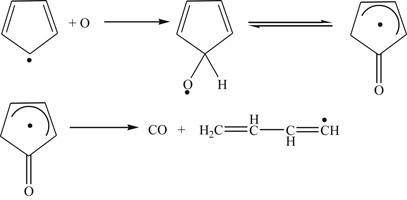 (3.151)
(3.151)The butadienyl radical found in reaction (3.151) then decays along various paths [61], but most likely follows path (c) of reaction (3.152):
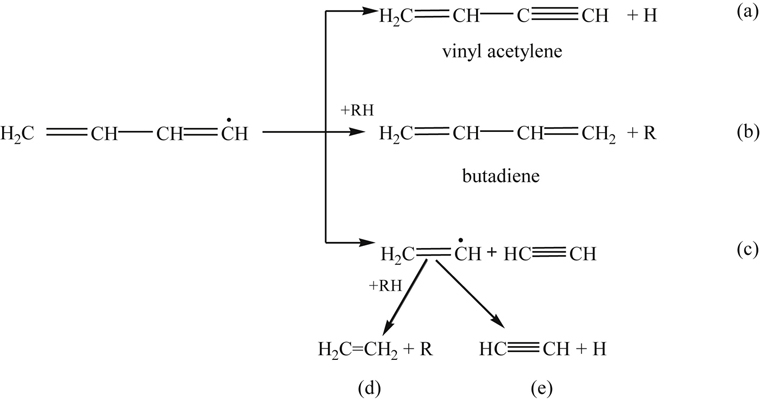 (3.152)
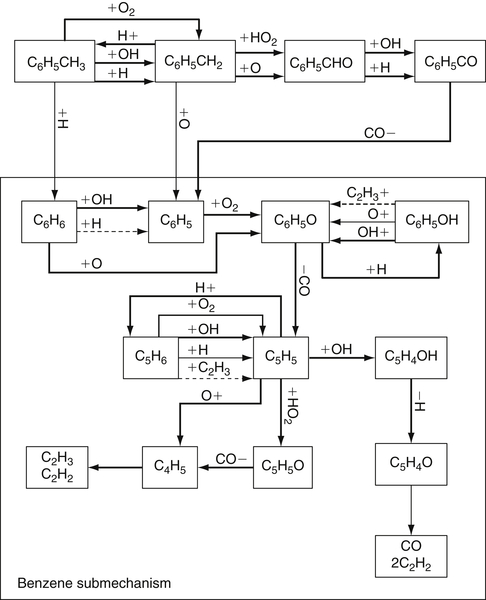
(3.152)Although no reported work is available on vinylacetylene oxidation, oxidation by O would probably lead primarily to the formation of CO, H2, and acetylene (via an intermediate methyl acetylene) [51]. The oxidation of vinylacetylene, or the cyclopentadienyl radical shown earlier, requires the formation of an adduct (as shown in reaction (3.151)). When OH forms the adduct, the reaction is so exothermic that it drives the system back to the initial reacting species. Thus, O atoms become the primary oxidizing species in the reaction steps. This factor may explain why the fuel decay and intermediate species formed in rich and lean oxidation experiments follow the same trend, although rich experiments show much slower rates [94] because the concentrations of oxygen atoms are lower. Figure 3.14 is a summary of the reaction steps that form the general mechanism of benzene and the phenyl radical oxidation based on a modified version of a model proposed by Emdee et al. [90,95]. Other models of benzene oxidation [96,97], which are based on Ref. [90], place emphasis on different reactions.
3.8.3.2. Oxidation of alkylated aromatics
The initiation step in the high-temperature oxidation of toluene is the pyrolytic cleavage of a hydrogen atom from the methyl side chain, and at lower temperatures it is O2 abstraction of an H from the side chain, namely,
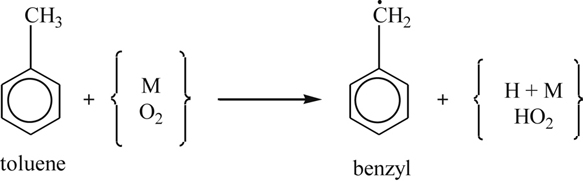 (3.153)
(3.153)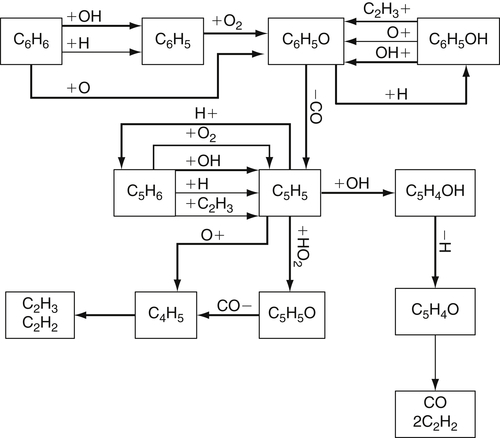
The H2–O2 radical pool that then develops begins the reactions that cause the fuel concentration to decay. The most effective attackers of the methyl side chain of toluene are OH and H. The OH radical does not add to the ring, but rather abstracts an H from the methyl side chain. This side-chain H is called a benzylic H. The attacking H has been found not only to abstract the benzylic H but also to displace the methyl group to form benzene and a methyl radical [98]. The reactions are then
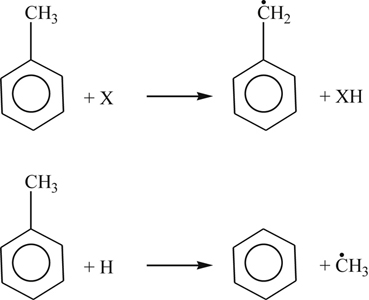 (3.154)
(3.154)The early appearance of noticeable dibenzyl quantities in flow reactor studies certainly indicates that significant paths to benzyl exist and that benzyl is a stable radical intermediate.
 (3.155)
(3.155)The reaction of benzyl radicals with O2 through an intermediate adduct may not be possible, as was found for reaction of methyl radical and O2. (Indeed, one may think of benzyl as a methyl radical with one H replaced by a phenyl group.) However, it is to be noted that the reaction
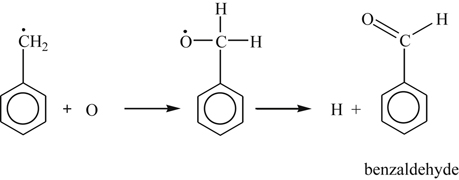 (3.156)
(3.156)has been shown [47] to be orders of magnitude faster than reaction (3.155). The fate of benzaldehyde is the same as that of any aldehyde in an oxidizing system, as shown by the following reactions that lead to phenyl radicals and CO:
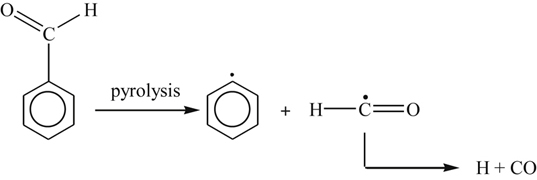 (3.157)
(3.157)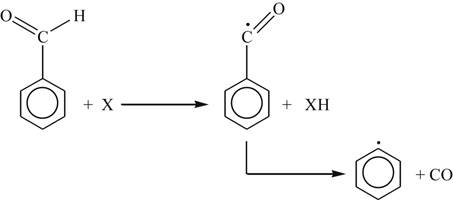 (3.158)
(3.158)Reaction (3.158) is considered the major channel.
Reaction (3.156) is the dominant means of oxidizing benzyl radicals. It is a slow step, so the oxidation of toluene is overall slower than that of benzene, even though the induction period for toluene is shorter. The oxidation of the phenyl radical has been discussed, so one can complete the mechanism of the oxidation of toluene by referring to that section. Figure 3.15 from Ref. [95] is an appropriate summary of the reactions.
The first step of other high-order alkylated aromatics proceeds through pyrolytic cleavage of a C–C bond. The radicals formed soon decay to give H atoms that initiate the H2–O2 radical pool. The decay of the initial fuel is dominated by radical attack by OH and H, or possibly O and HO2, which abstract an H from the side chain. The benzylic H atoms (those attached to the carbon next to the ring) are somewhat easier to remove because of their lower bond strength. To some degree, the benzylic H atoms resemble tertiary or even aldehydic H atoms. As in the case of abstraction from these two latter sites, the case of abstraction of a single benzylic H can be quickly overwhelmed by the cumulative effect of a greater number of primary and secondary H atoms. The abstraction of a benzylic H creates a radical such as
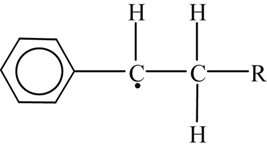
which by the β-scission rule decays to styrene and a radical [84]
 (3.159)
(3.159)where R can, of course, be H if the initial aromatic is ethyl benzene. It is interesting that in the case of ethyl benzene, abstraction of a primary H could also lead to styrene. Apparently, two approximately equally important processes occur during the oxidation of styrene. One is oxidative attack on the double-bonded side chain, most probably through O atom attacks in much the same manner that ethylene is oxidized. This direct oxidation of the vinyl side chain of styrene leads to a benzyl radical and probably a formyl radical. The other is the side-chain displacement by H to form benzene and a vinyl radical. Indeed the displacement of the ethyl side chain by the same process has been found to be a major decomposition route for the parent fuel molecule.
If the side chain is in an “iso” form, a more complex aromatic olefin forms. Isopropyl benzene leads to a methyl styrene and styrene [99]. The long-chain alkylate aromatics decay to styrene, phenyl, benzyl, benzene, and alkyl fragments. The oxidation processes of the xylenes follow somewhat similar mechanisms [100,101].
3.8.4. Supercritical Effects
The subject of chemical reactions under supercritical conditions is well outside the scope of matters of major concern to combustion-related considerations. However, a trend to increase the compression ratio of some turbojet engines has raised concerns that the fuel injection line to the combustion chamber could place the fuel in a supercritical state; that is, the pyrolysis of the fuel in a flow line could increase the possibility of carbon formation as soot. The question then arises as to whether the pyrolysis of the fuel in the line could lead to the formation of PAH, which generally arise in the chemistry of soot formation (see Chapter 8, Section 8.5.6). Since the general conditions in devices of concern are not near the critical point, then what is important in something like a hydrocarbon decomposition process is whether the high density of the fuel concentrate affects the decomposition kinetic process so that species would appear other than those that would occur in a subcritical atmosphere.
What follows is an attempt to give some insight into a problem that could arise in some cases related to combustion kinetics, but not necessarily related to the complete field of supercritical use as described in pure chemistry texts and papers. It is apparent that the high pressure in the supercritical regime affects not only the density (concentration) of the reactions, but also the diffusivity of the species that form during pyrolysis of important intermediates that occur in fuel pyrolysis. Indeed, as well, in considering the supercritical regime, one must also be concerned that the normal state equation may not hold.
For example, in some early work on the high-temperature pyrolysis of the endothermic fuel methylcyclohexane (MCH), it was found that in the subcritical state MCH pyrolysis is β-scission dominated (see Section 3.8.1) and little, if any, PAH are found [102,103]. What is of interest is that it was found that while β-scission processes are still important under supercritical conditions, they can be significantly slower [104]. These studies suggest that the pyrolysis reaction of MCH proceeds to form the methylhexedienly radical (MHL) under both sub- and supercritical conditions [102,103]. However, under the supercritical condition it was found that dimethylcyclopentane subsequently forms. The process by which the initial six-member ring is converted to a five-member ring is most apparently due to the phenomenon of caging, a phenomenon frequently discussed in the supercritical chemical process literature [104]. The formation of a cyclic intermediate is more likely to be PAH. Thus, once MHL forms, it can follow two further possible routes: β-scission leading to ordinary pyrolysis or a cyclization due to the phenomenon called caging. The extent of either depends on the physical parameters of the experiment, essentially the density (or pressure). A graphical presentation of this item is given in Figure 3.16.
In order to estimate the effect of caging with respect to a chemical reaction process, the general approach has been to apply transition state theory [104]. What has been considered in general transition state theory (see Chapter 2, Section 2.2.2) is the rate of formation of a product through an intermediate (complex) in competition with the intermediate reforming the initial reactant. Thus, β-scission is considered in competition with caging. However, in essence, the preceding paragraph extends the transition state concept in that the intermediate does not proceed back to the reactants, but has two possible routes to form different products. One route is a β-scission route to produce general hydrocarbon pyrolysis products and the other is a caging process possibly leading to a product that can cause fuel line fouling in a way as illustrated in Figure 3.16.

Following a general chemical approach [104] to evaluate the extent of a given route, it is possible to conclude that under supercritical conditions, the extent of fuel fouling (PAH formation) could be determined by the ratio of the collision rate of formation of the new cyclohydrocarbon due to caging to the diffusion rate of the β-scission products “to get out of the cage.” This ratio can be represented by the expression [νd2 exp(−E/RT)/D] or [ν exp(−E/RT)/(D/d2)], where ν is the collision frequency (s−1), d2 the collision cross-section, E the activation energy, and D the mass diffusivity (cm2/s) [104]. The second ratio expression will be recognized as a Damkohler number [104]. For the pyrolysis process referred to, the caging institutes a bond formation process, and thus activation energy does not exist. Then the relevant Damkohler number is [ν/(D/d2)].
Typical small molecule diffusivities have been reported to range from 10−1 cm2/s for gases to 10−5 cm2/s for liquids [105]. One would estimate that under supercritical conditions the supercritical fluid would be somewhere between the two values. It has been proposed that although supercritical fluids have in many instances greater similarity to liquids than gases, their diffusivities are more like gases than liquids. Thus, the caging product should increase with pressure, as has been found in MCH pyrolysis [103] and, perhaps, in other similar cases related to combustion problems.
3.8.5. Biofuels
Biofuels are fuels that contain energy from geologically recent carbon fixation. The two most common types of biofuels are small-molecule alcohols (such as ethanol and butanol) and large-molecule biodiesel fuels. Alcohols are commonly made by fermentation, mostly from biomass high in carbohydrates, that is, sugar and starch crops, while biodiesel is produced from vegetable oils or fats using transesterification to yield fatty acid methyl esters (FAMEs) or fatty acid esters.
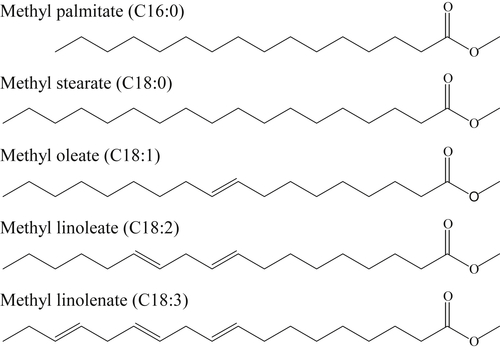
Biodiesel fuels derived from soybean or rapeseed generally consist of a limited number of FAMEs that include methyl stearate, methyl oleate, methyl linoleate, methyl linolenate, and methyl palmitate (Figure 3.17), which include one, two, or three CC double bonds. Characteristic of biodiesel fuels is also the presence of the methyl ester group at one end of the carbon chain, which contains two oxygen atoms. Several recent reviews of biofuel combustion are available [106–109].
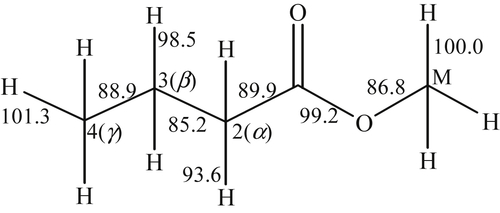
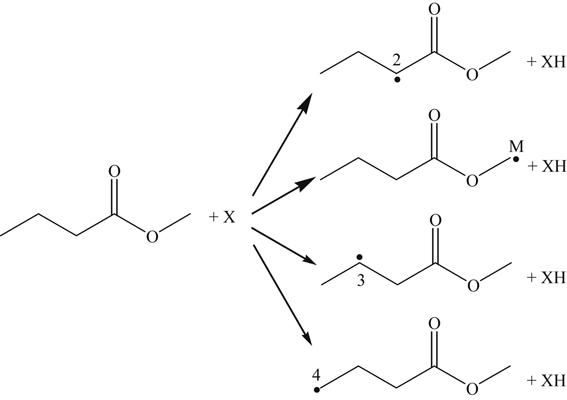
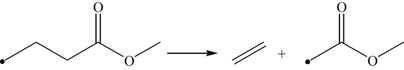
Methyl butanoate is a relatively small methyl ester (Figure 3.18), which received early study because of its size and the fact that it possesses several essential features of larger FAMEs [107,110]. Included in Figure 3.18 are the methyl butanoate bond energies reported by Dooley et al. [110] showing the C–H bond α to the carbonyl group as the weakest C–H bond. Like the oxidation of hydrocarbons considered earlier, the reaction is initiated at intermediate temperatures through H atom abstraction by molecular oxygen and at high temperatures via unimolecular fuel decomposition by CC (C2H5–CH2C(O)OCH3 and n-C3H7C(O)O–CH3) and C–H bond scission. At high temperatures, once a radical is formed, the reaction proceeds mainly through hydrogen abstraction at the 2, M, 3, and 4 positions (in decreasing order of importance) according to
 (3.160)
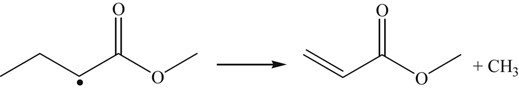
(3.160)where X is OH, H, O, CH3, and HO2. The primary fuel radical formed is by H atom abstraction at the 2 position, which undergoes β-scission to form an alkyl radical (methyl) and an oxygenate.
 (3.161)
(3.161)Further radical attack of the oxygenate and β-scission yields formaldehyde, carbon monoxide, and the vinyl radical.
 (3.162)
(3.162)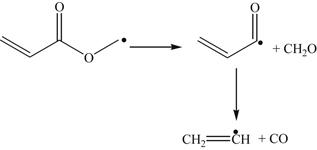 (3.163)
(3.163)Hydrogen abstraction at the carbon M position is of next importance, which after β-scission yields formaldehyde, carbon monoxide, and the n-propyl radical.
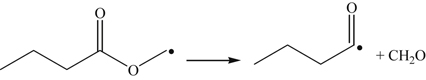 (3.164)
(3.164)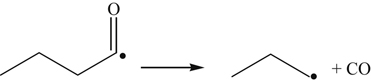 (3.165)
(3.165)Decomposition of the fuel radical formed by H abstraction at the β or carbon 3 position yields the methoxy formyl radical (CH3CO), which is important to early CO2 formation in methyl butanoate combustion.
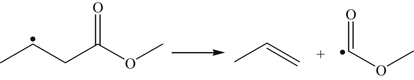 (3.166)
(3.166)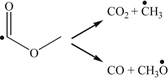 (3.167)
(3.167)Evident from the reaction scheme described, the reaction generally leads to parallel paths of oxidizing the methyl ester group and the alkyl radicals formed after the initial β-scission. Thus, in the case of large methyl esters, which have similar ordering of bond energies, it is not surprising that the high-temperature reactivity has similarity to large n-alkanes [111]. At low temperatures, the resulting alkyl radicals of methyl butanoate are too small to involve low-temperature “cool-flame” chemistry with negative temperature coefficient behavior; the degree of low-temperature chemistry for larger methyl esters depends upon the molecular structure [112].
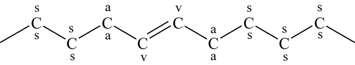
An important difference between biofuels and most petroleum-based hydrocarbon fuels as noted by Westbrook [106] is the number of fuel species with CC double bonds. Most biofuels contain fuel species with one or more CC double bonds as shown in Figure 3.17, whereas the important bonds in conventional hydrocarbon fuels are CC single bonds. The importance of this structural characteristic has a significant influence on low-temperature chemistry. The influence is most readily observed in the cetane number of the various methyl esters of Figure 3.17. The cetane number is a measure of the ignitability of a fuel; that is, the larger the cetane number, the easier it is to ignite the fuel. Cetane numbers for methyl stearate, methyl oleate, methyl linoleate, and methyl linolenate are 101, 59, 38, and 23, respectively, showing that increasing the number of CC bonds clearly reduces the cetane number, or more importantly, the rates of reaction leading to ignition. Westbrook [106] explains this trend through the C–H bond strengths adjacent to the doubly bonded carbon atoms. Considering a portion of methyl oleate with one CC double bond (3.168) and the same portion of methyl linoleate, but with two CC double bonds (3.169),
 (3.168)
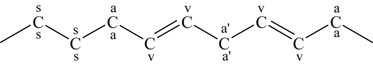
(3.168)identifies the different types of CH bonds of interest, that is, those at the allylic sites (indicated with the letter a), those at the secondary sites (indicated by the letter s), and those of the bis-allylic methylene group (indicated by the letter a'), which are located between the two CC double bonds of methyl linoleate.
 (3.169)
(3.169)The CH bonds at the bis-allylic sites are weaker than those at the allylic sites (∼367 kJ/mol or 87.8 kcal/mol), which are significantly weaker than those at the secondary sites (∼410 kJ/mol or 98 kcal/mol). Thus, preferential abstraction of weakly bonded hydrogen atoms produces radicals at the allylic sites of methyl oleate and the bis-allylic sites of methyl linoleate. The vinylic CH bonds, which are the H atoms bonded to the C atoms in the double bond (indicated by the letter v) are considerably stronger (∼452 kJ/mol or 108 kcal/mol) than the secondary CH bonds and need not be considered. At low temperatures (below ∼900 K), the allylic and bis-allylic radical sites represent the locations for O2 addition and the formation of the alkylperoxy radicals that can undergo isomerization and produce the low-temperature chemistry of Figure 3.11. However, just as noted for the CH bonds at these two sites, the R–O2 bond energies are considerably less than at the ordinary secondary sites. Westbrook et al. [111] determined the bond dissociation energies for all possible R–O2 bonds in methyl stearate, methyl oleate, methyl linoleate, and methyl linolenate. Their results indicate that the R–O2 bond energies are approximately 159 (38), 113 (27), and 67 (16) kJ/mol (kcal/mol) at the secondary, allylic, and bis-allylic sites, respectively. Thus, the weakly bound O2 means that the R–O2 almost immediately decomposes back to reactants, and hence very little low-temperature chemistry is initiated at these sites because of the short lifetime of the adduct. Consequently, biofuel species with more CC double bonds react more slowly at low temperatures.
At low temperatures, O2 addition to the hydrocarboxyl radical (OCHO) may also occur and lead to another route for early carbon dioxide formation. Herbinet et al. [113] proposed isomerizations of the peroxy radicals formed from the methyl ester group. For methyl butanoate, hydrogen abstraction at the carbon 4 site (reaction (3.160)) followed by β-scission yields
 (3.170)
(3.170)Oxygen addition with subsequent isomerization produces a cyclic ether.
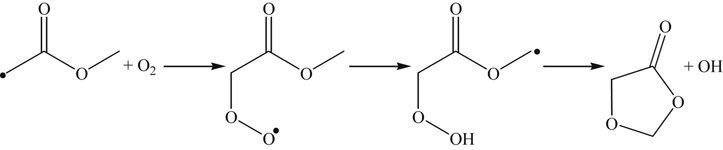 (3.171)
(3.171)Hydrogen abstraction of the cyclic ether followed by the opening of the ring leads to early CO2 formation as shown below.
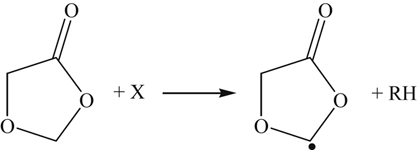 (3.172)
(3.172) (3.173)
(3.173) (3.174)
(3.174)The above sequence occurs in large FAMEs as well because breakage of C–O bonds are generally energetically unfavorable compared to C–C bonds. For example, Osmont et al. [114,115] determined the lowest bond dissociation energy in methyl palmitate for a C–O bond was ∼372 kJ/mol (89 kcal/mol), while the lowest bond dissociation energy for a C–C bond was ∼347 kJ/mol (83 kcal/mol). Thus, the carbon and oxygen remain bonded throughout the combustion process. The size of the transition state (for example, reaction (3.171)) has a large influence on the reaction barrier, and hence on whether other isomerization pathways are preferred energetically to early CO2 formation [116].
In addition to the reaction pathways discussed here, the presence of oxygen in biofuels also has important implications on the sooting behavior of flames as discussed in Chapter 8.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.