
Chapter 5
Photo-Stimulated Phase Transformations in Liquid Crystals and Their Non-Display Applications
5.1 Introduction
The anisotropy of molecular shape is the prime factor responsible for the formation of liquid crystalline phases [1–3]. Manipulating this anisotropy by altering the chemical structure of the molecules has been the traditional way of stabilizing/destabilizing liquid crystal (LC) phases [4–7]. While this has been very efficient and has resulted in a wide variety of organic molecules exhibiting rich mesomorphic behavior, the possibility to control the anisotropy by a non-chemical external means has always been attractive. The modification in the shape of the molecules owing to conformational change driven by photoisomerization of certain systems responsive to light of a particular wavelength, called actinic light, has served this need very well. Photochemical pathways have the advantage over thermal and catalytic methods of giving mixtures rich in energetically unstable isomers. Photoisomerization is a photochemical process leading to an isomerization of a certain chemical entity, either by bond rotation, skeletal rearrangement, or atom/group-transfer [8] in chemical compounds. A number of organic compounds such as azobenzenes, stilbenes, alkenes are good examples of photoisomerizable molecular systems, which are also called photochromic/photoactive/photoresponsive compounds.
The organization of the chapter is as follows. We start with a brief introduction to the phenomenon of photoisomerization [8–20]. Section 2 gives a survey of the diverse photoinduced phase transitions (PIPT) observed in liquid crystalline systems involving achiral as well as chiral materials, nematic, smectic, columnar, and blue phases; a detailed account of certain unique behavior of these photo-stimulated transformations [21–26, 28–30, 36–38, 41, 42, 46–48, 53, 54, 59, 60, 63–89, 101–211] is provided in Section 3. The chapter concludes with a note on two specific non-display applications based on the ability of light to induce large changes in the properties of liquid crystals, and which have attracted tremendous attention [27, 31–35, 39, 40, 43–45, 49–52, 55–58, 61, 62, 90–100, 212–219].
5.2 Survey of Photoinduced Phase Transformation in Liquid Crystals
Over the last four decades, a number of light-driven isothermal phase transitions have been reported in LCs due to reversible structural change in the constituent photoresponsive component(s). Among a variety of photoresponsive systems known, azobenzenes, viz., azobenzene and its derivatives, have been extensively used to bring about such phase transitions. This is not only because of the ability of azobenzenes to exhibit fast, efficient, and fully reversible photoisomerization, but also their isomerization is one of the cleanest photoreactions ever known where side-products are not formed even with innumerable isomerization cycles [9–12]. For these reasons and also for the fact that majority of the results presented in this chapter employ azobenzenes to bring about photo-driven effects, we mainly consider the studies involving azobenzenes for the following discussions.
5.2.1 Photoisomerization
The elucidation of the isomerization mechanism in azobenzene has been substantially debated in the literature [13–20] and the latest semiclassical dynamics simulation suggests that the photoisomerization of trans-azobenzene follows an inversion-assisted rotation path [20]. Azobenzene may exist as two geometric isomers, the trans and the cis form, also referred to as E and Z forms, respectively. These two configurations differ in the direction of the central bonds as shown in Figure 5.1. In the energetically more stable E state the two bonds linking the azobenzene group to the aromatic rings are parallel, resulting in an elongated shape of the molecule. In the metastable Z state the molecules adopt a bent conformation. The two isomers also differ in their absorption spectra. The absorption spectrum of E-azobenzene exhibits a maximum at around 350 nm due to the π–π* electronic transition and another at around 440 nm due to the n–π* transition (Figure 5.2). The attractive feature, at least from the point of view of the present studies, is that the two forms are inter-convertible by the use of actinic light. Typically the absorption of one light quantum of radiation in the UV region (~350 nm) results in the E → Z transformation accompanied by changes in electronic spectra of the material, the phenomenon being called photochromism. On the other hand, the Z isomer can be converted back to the E isomer by shining visible light (~440 nm). Since the E isomer is energetically more stable than the Z isomer, the back conversion also occurs spontaneously, through a process known as thermal back relaxation (TBR). Temperature, host environment, type of dye, and excitation wavelength are among the parameters, which play a decisive role in the character of photoisomerization.
Figure 5.1 A facile photoinduced geometric isomerization of azobenzene about the azo bond, converting the trans (E) molecule into cis (Z) isomer. Note that the cis-isomer thermally reverts to the stable trans state. Also shown are the cylindrical and space filling models of energy minimized E and Z forms of azobenzene molecule to signify extremely large changes in conformation (shape), size (length), and molecular dipole (polarity).
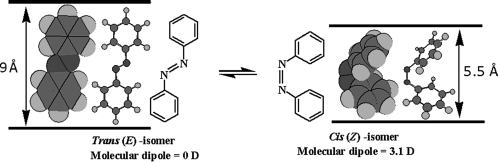
Figure 5.2 Molar absorptivity (εM) spectra for a typical azobenzene, EPH, and its mixture (5 wt%) with a nematic LC obtained using 0.1 mM/l solutions with 1,4-dioxane as a solvent. The peaks around 360 nm correspond to the π–π* transition of the trans isomer of EPH. The higher wavelength (~450 nm) peak in both the materials is due to the n–π* transition of the cis isomer of EPH. The 280 nm peak seen for the mixture arises from the biphenyl moieties present in the parent LC, as confirmed by its spectrum. The inset shows the effect of photoisomerization on the relative concentration of trans and cis isomers, with the absorption for the former decreasing (indicated by down arrow), and the latter increasing (up arrow) [ref. 179].

5.2.2 PIPT and Related Phenomena
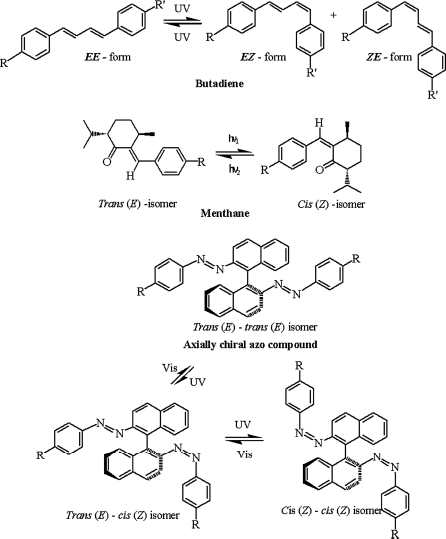
LC phase transitions are generally known to come about when the external parameters such as temperature, concentration, and pressure are varied. However, in recent years, irradiation with light of suitable wavelength—the photon action—has been recognized as another important stimulus that induces LC phase transitions [21–27]. In fact, it has been proven that actinic light serve as a fascinating tool to study condensed matter from a new dimension where the LC phases are stabilized/modulated through the photoisomerization of the constituent photoisomerizable molecules. From this viewpoint, the most important property that changes as a result of the photo-driven isomerization is the shape (geometry/conformation) of the molecule [see for example, refs. 9, 14, 19, 20]. Several extensive review articles on the topic of photoinduced phase transition (PIPT) in LCs exist in the literature [21–27]. Such light-driven transitions and closely related phenomena have been experimentally demonstrated since 1970s [28–34] where either photochromic molecules which themselves are mesomorphic or photoactive guest–LC host composites have been used [21–88, 180]. In general, apart from azobenzenes, other photoresponsive molecules such as stilbenes [29, 30, 63–65], azoxybenzenes [66–68], spiropyrans [63–65, 69], fulgides [63–65, 70, 71], diarylethenes [63–65, 72–78, 211], azabipyridines [79], butadienes [80, 81, 134, 135, 144, 155], menthanes [82, 83], and axially chiral azo compounds [65, 84–88] have been utilized to generate photoresponsiveness in LCs. Chart 5.1A and B depict the molecular structures of such photoactive organic compounds and their respective isomers/products formed after irradiation with actinic light.
Chart 5.1A Molecular structures of photoactive compounds and their isomers/products formed after their illumination with light of suitable wavelength [refs. 21–23, 27, 63–65].
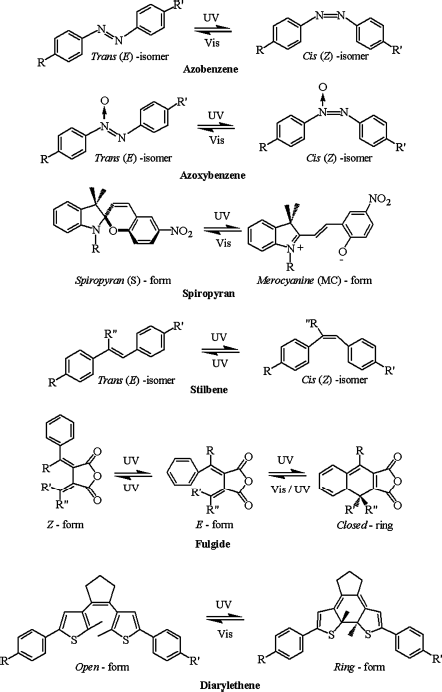
Chart 5.1B Molecular structures of photoactive compounds and their isomers/products formed after their illumination with light of suitable wavelength [refs. 21–23, 27, 63–65].
As mentioned earlier, of all the known photoactive systems, azobenzene and its derivatives have attracted by far the most attention; specifically, besides being used as a dopant, the azobenzene core has been utilized in the design and synthesis of both low molar mass and polymeric LCs. The approach wherein azobenzene (or its derivative) is employed as a dopant/one of the pendant moieties in a copolymer, provides an invaluable opportunity to design different guest–host combinations, and thereby to engineer (at least to some extent) various properties such as order parameter, birefringence, color, viscosity, and polarity of the resulting photoresponsive system. In the following, we discuss about the various general aspects of photoinduced effects on LCs with special emphasis on phase transitions and related phenomena.
5.2.3 Photoinduced Nematic to Isotropic Phase Transition
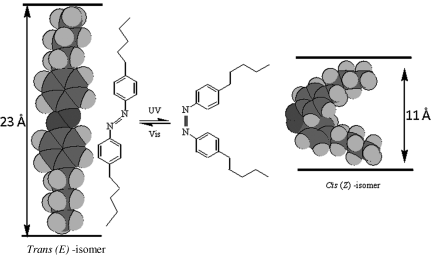
Photoinduced phase transition resulting in the disordering of the nematic (N) LCs has attracted much attention from both fundamental and technology viewpoints [21–27, 31–62, 95–103, 163, 167, 168, 171, 173–177, 179, 180, 212]. In particular, the nematic-to-isotropic (N-I) transition is one of the most well-studied photoinduced isothermal phase transitions, which is generally seen in systems derived from azobenzene [11–42]. As mentioned earlier, the photoactive azobenzene, after substituting with suitable groups, can itself be mesomorphic or chemically attached to the liquid crystal molecule or used as a dopant to a LC host material. In such systems, the E form of the azobenzene dopant, as it is rod-like, is favorable for the stabilization of the liquid crystalline phase. On the other hand, the reduced shape anisotropy and the length of Z form is less in favor of liquid crystallinity. For example, the length of the E isomer of 4,4′-dipentylazobenzene, deduced from MM2 computations of CS Chem Draw 3D (version 5) program, is about 2.3 nm, whereas the Z isomer is nearly half of it, being about 1.1 nm as shown in Figure 5.3. It is this combination of dramatic change in the shape and length of the molecule in association with the high photochemical stability of azobenzenes that lies at the heart of the phenomenon of photoinduced transitions. In fact, the Z form with a striking bent-shape acts like an “impurity” and therefore destabilizes the liquid crystalline phase. Hence, photoisomerization from the E to the Z form causes, in general, a lowering of the transition temperature. If, for instance, the material containing the photoactive azobenzene derivative exhibits a N-I transition and the UV irradiation is done in the nematic phase, the gradual lowering of the transition temperature (TNI), due to the accumulation of cis-isomers, could induce a transition to the isotropic phase. It must be emphasized that this transition is photochemical in nature and not driven by a change in temperature and therefore is an isothermal phase transition. Further, the photoinduced transitions in liquid crystalline materials generally employ one-photon excitation of the azobenzene group. However, recently, a photo-driven phase transition has been accomplished through two-photon excitation [89]. Such a two-photon driven trans–cis isomerization has the added advantage of controlling the location and spatial extent of a photo-stimulated structural distortion and expected to hold the promise for advanced opto-electronic and photonic applications.
Figure 5.3 Molecular structures and the space filling models of E–Z isomers of 4,4′-dipentylazobenzene. Note that the trans-isomer possess rod-like geometry while the cis-isomers attain bent-conformation and as a result the length, and the shape anisotropy of the molecule gets reduced considerably.
5.2.4 Characterization of Liquid Crystal to Isotropic Phase Transition Using POLARIZING OPTICAL MICROSCOPE
Given the fact that in contrast to the isotropic phase the LC phases have a finite birefringence, polarizing optical microscopy (POM) can be used as a convenient probe to visualize the photoinduced isothermal LC-I transition. An example of such an observation for a system exhibiting photo-driven nematic-to-isotropic transition (see the following section for details) is shown in Figure 5.4. The sample in the N phase placed between crossed polarizers was illuminated uniformly with UV radiation except in the central region. The region that received the radiation underwent the photoinduced isothermal transition and therefore appears dark indicating the isotropic phase while the central masked portion, which is unirradiated, stays in the birefringent nematic state. Liquid crystals are elastically soft materials. If a small portion of LC molecules changes its orientation in response to an external stimulus, the other LC molecules tend to follow the change. Upon photoisomerization, the induced bent form of the photoactive molecules introduces a local region of higher orientational entropy (disorder) compared to the regions of the host (rod-like non-photoactive) molecules. If the generated disorder is large enough, the formation of the isotropic phase domains at the irradiated sites appears as dark regions under the microscope. The sphere of influence of the azobenzene molecules could become very large making the entire sample undergo the photoinduced transition from the nematic to the isotropic phase. It may be noted that the boundaries of the phases are quite sharp (except for a slight birefringence change between the middle and the edges, owing to slight light leakage) and if not illuminated with 450 nm light, it remains so until the thermal back relaxation occurs. A question that arises is why the boundaries should be sharp? The sample is a fluid and therefore why the azobenzene molecules converted into their cis state by UV light cannot disperse uniformly and results in an overall reduction of the order parameter rather than transforming the illuminated region to the isotropic phase? The reason must be the nematic potential. The molecules in regions containing the Z isomers and having undergone the transition to the nematic state should climb the nematic potential to reach the unirradiated region. The potential produces a barrier, which prevents an easy diffusion, thus creating a sharp boundary between the illuminated and non-illuminated regions.
Figure 5.4 Photomicrograph demonstrating the isothermal nematic–isotropic transition driven by UV light.

The accumulated results indicate that the critical intensity at which the N-I transition occurs depends on several factors including the chemical nature of photoactive molecules [21–62, 180]. In fact, this was indicated in one of the earlier reports on photo-driven N-I phase transition [32] where a small amount of azobenzene or its derivative, 4-n-butyl-4′-methoxyazobenzene (BMAB), was doped in a standard nematic LC, 4-cyano-4′-n-pentylbiphenyl (5CB) (see Chart 5.2). In these binary mixtures showing photo-driven N-I transition, the trans–cis isomerization was found to be nearly linear with the reaction time. A parameter termed as amplification by the authors was better for BMAB than for the unsubstituted azobenzene. In the last four decades a number of single component low molar mass/polymeric and guest–host nematic LCs exhibiting photoinduced N-I transition have been investigated with a view to understand the structure–property correlations. In fact, the odd–even feature exemplified in dimers has been recently highlighted for the purpose of examining photoinduced effects [26, 139–144, 177]. In a later section we point out how the length and parity of the spacer is a powerful control parameter to alter the behavior of the N-I transition specifically.
Chart 5.2 Molecular structures of nematic host LC (5CB), photoactive LC monomeric azobenzenes (BMAB and 8AB8), and photoactive LC side-chain polymers (PA6AB2, PM6AB2, PA6ABCN, and PM6ABCN) used in early important studies [refs. 32 and 35].
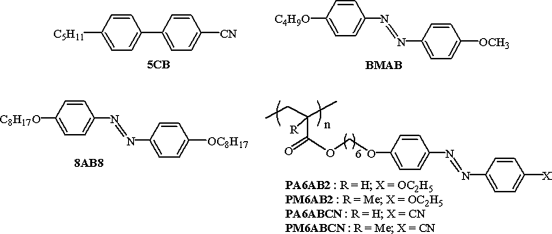
Thus, it is quite apparent that different azobenzene derivatives have been used in accomplishing the photoinduced N-I transition. However, azoxybenzene compounds, which are stable and exhibit mesomorphism analogous to azobenzenes, have also been demonstrated to be effective for such photoinduced transitions [66–68]. One such study [68] involved a room temperature nematic LC made of 4-n-butyl-4′-methoxyazoxy-benzene and 4-n-butyl-4′-heptanoylazoxybenzene. The results revealed that in less than a minute a moderate UV intensity can bring about trans–cis isomerization with nearly equal amounts of both isomers present. It was also observed that a cis concentration of 12% is enough to lower the N-I transition from 70°C to room temperature highlighting the importance of azoxybenzene compounds. As we shall discuss briefly in the concluding section, photoinduced N-I transition has been well used in optical storage (holography) applications [31–35, 39, 40, 43–45, 49–52, 55–58, 61, 62, 212] as well as in devising soft actuators [27, 90–103, 213–219].
5.2.5 Photoinduced Nematic to Chiral Nematic Phase Transition
Appearance of chiral order in achiral materials has always been fascinating [104, 105]. In LCs, an entire class of materials, referred as banana mesophases, has seen an enormous activity owing to such a feature [104, 106–109]. In general several approaches to realize absolute asymmetric synthesis employing a chiral stimulus are known [110, 111]. Especially attractive, from the present context, is the usage of circularly polarized light (CPL) for this purpose. In fact, the conversion between achiral nematic (N) to chiral nematic (N*) phases by CPL in guest–host composites has been achieved, where the guest systems (dopants) are racemic photochemical organic compounds [63–65, 111–117]. Indeed, the initial work on reversible photoresolution of metal-complexes [118–121] and bridgehead imine [122] made it possible to use this novel phenomenon in the LC field with the early demonstration by Suarez and Schuster in 1995 [112]. For realizing N-N* transition, a photoactive racemic compound, referred to as “racemic (optical) trigger”, is dissolved in the host nematic LC and the system is irradiated with CPL, resulting in enantiodifferentiation as enantiomers of the racemic dopant absorb CPL with different probability. For example, Huck et al. [116] demonstrated the transition from N to N* phase using the overcrowded racemic alkene [116] in a nematic host, 4′-(pentyloxy)-4-biphenylcarbonitrile (5OCB) and irradiation by CPL at 313 nm (see Figure 5.5). It was noted that the transition was due to the chiral enrichment of the enantiomer (M)-alkene, and that the handedness of the helix of the N* phase directly corresponds to that of the dopant. Restoration to the racemic nematic state was obtained upon illumination with unpolarized light.
Figure 5.5 Photoinduced effects on an overcrowded alkene optical trigger dissolved in a nematic LC (5OCB): (a or b) resolution of racemic alkene into (P)-alkene or (M)-alkene and vice versa (racemization); (c) interconversion of enantiomeric (P) and (M) forms [ref. 116]. Here, the N-N* transition is achieved by illuminating the N phase of a binary system composed of a guest photoactive racemic compound, and the host nematic LC; the reverse (N*-N) phase transition is accomplished by illuminating the N* phase with the unpolarized light. (LPL: linearly polarized light; UPL: unpolarized light; r-CPL: right handed-circularly polarized light; l-CPL: left handed-circularly polarized light).
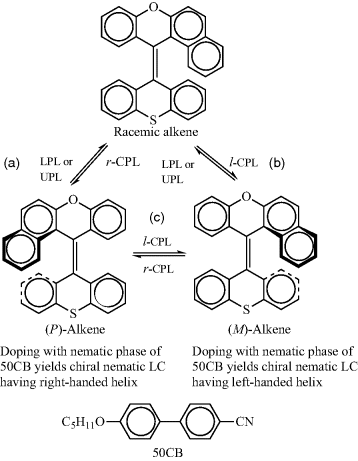
The reverse N*-N phase transition is achieved by photoracemization caused by illumination with unpolarized light. As mentioned above, Suarez and Schuster [112] used a chiral bicyclic ketone substituted with an exocyclic double bond as a dopant in a nematic LC namely, trans-1-n-heptyl-4-(p-cyano)phenylcyclohexane (Chart 5.3), to generate the N* phase. Irradiation of the sample causes racemization of the chiral dopant. As the concentration of racemic component increases (with increasing irradiation), the helical pitch of the N* phase increases, and finally it transforms to the N phase. Likewise, a photoswitchable donor–acceptor substituted, inherently dissymmetric compounds, (M)-alkene-1 and (P)-alkene-2 (Figure 5.6) have also been shown to be very effective chiral guest molecules in the light-assisted reversible N*-N phase transitions [115]. It may be worth noting that the enantiodifferentiation has been observed in dimers [123, 124], bent-core molecules [125], columnar mesomorphs [126], and polymeric materials [127–129].
Chart 5.3 Molecular structures of the guest optically active bicyclic ketone and the host nematic LC, trans-1-n-heptyl-4-(p-cyano)phenylcyclohexane used to demonstrate the N*-N phase transition [ref. 112].
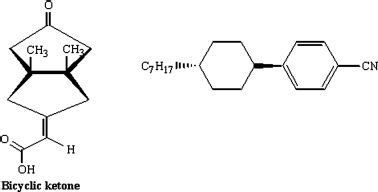
Figure 5.6 Light-assisted interconversion of (M)-alkene-1 and (P)-alkene-2; the N*-N transition is achieved by irradiating the N* phase of a binary system consisting of a guest (M)-alkene-1 and the host nematic LC 5OCB; the N* phase is regained if the N phase is illuminated with 435 nm light [ref. 115].

5.2.6 Photoinduced Transitions Involving Smectic and Blue Phases
Analogous to the aforementioned reversible I-N and N-N* transitions, a number of other remarkable photoinduced phase transitions involving interconversion of LC phases or their transformation to isotropic phase have been observed in a variety of systems. Yet again, the azobenzene chromophore has been incorporated in majority of the photo-functional materials. The observed phase transitions are smectic A (SmA)-to-isotropic (I) (SmA-I); SmA-N; SmA-to-crystal (Cr) state (SmA-Cr); SmA-N*; SmA-blue phase (BP) (SmA-BP); blue phase I (BP-I)-to-blue phase II (BPII) (BPI-BPII); BPII-to-blue phase III (BPII-BPIII); BPIII-I; N*-BPII; N*-BPI/BPII; smectic Q (SmQ)-to-SmQ (SmQ-SmQ); SmQ-I; chiral smectic C (SmC*)-to-I (SmC*-I); N*-I; SmC*-SmA; SmA-N*; SmC*-N*; columnar (Col)-to-I (Col-I). First we give an overview of photo-stimulated transformation of these sequences and then describe some specific examples in detail.
As is well known, the photolysis of photoactive LCs suppresses their clearing as well as LC–LC transition temperatures due to light-induced disordering of the system [21–26]; in fact, in certain cases, even the intensity of the microscope lamp source is sufficient to bring about such transitions [130–132]. Examples of photoactive compounds [131, 132] showing such effects and presenting the mesophases SmA, SmC*, twist grain boundary (TGB), N* and BP are given in Chart 5.4A. The photo illumination–temperature phase diagram was presented to show effect on, for example, the SmC*-N*-I phase sequence. The shifts in the LC–LC phase transition temperatures are found to depend on the imposed illumination and the sample thickness; this is obvious given the fact that the rate of conversion from trans to cis isomers depends on the penetration depth of the incident light. Besides, the photo-modulation of the spontaneous polarization of the SmC* phase, has been established for some of these compounds [131]. Further, in one of such dimers (18A in Chart 5.4A), a continuous decrease in SmC* to TGBA (TGB phase having SmA blocks) phase transition temperature with the increase in the UV light intensity has been observed recently [133].
Chart 5.4 (A) General molecular structure of some optically active azobenzene-based monomeric LCs employed to study light-induced effect on (i) LC phase sequences involving SmA, SmC*, TGB, BP, and N* phases and (ii) spontaneous polarization of the SmC* phase [refs. [131]–133]. (B) General molecular structure of supramolecular LC motifs formed by 4,4-bipyridine and alkoxybenzoic acids through H-bond association exhibiting photoinduced SmA-Cr transition [refs. 22, 79]. (C) Molecular structure of a chiral LC derived from 1,4-diphenylbutadiene core showing photoassisted SmA-BPI and SmA-N* transitions [ref. 135]. (D) Molecular structure of an optically active azobenzene-based atypical LC exhibiting reversible photoinduced SmQL-SmQH and SmQH-I transitions [ref. 138].

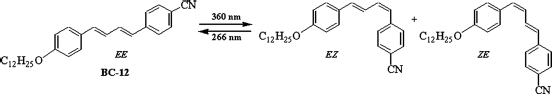
Light-stimulated SmA-I transition has been observed in a monomeric rod-like LC BC-12 (Fig. 5.7) where the diphenylbutadiene core is substituted with a cyano group and a dodecyloxy tail [134]. A thin film of SmA phase, upon irradiation with 360 nm light at 120°C, transforms into an isotropic liquid state. This process was found to be thermally irreversible; that is, the isotropic state directly transforms into the crystalline state. The experimental results evidence that the photoinduced SmA-I transition is due to the conversion of trans (EE) isomers to the cis (EZ and ZE) isomers (Figure 5.7). Interestingly, irradiation at room temperature of the photoinduced isotropic phase with 266 nm laser, results in the generation of the system exhibiting the thermal behavior analogous to that of the EE isomers of BC-12. Such a transition has also been observed in photoactive chiral dimers which we discuss in a later section. It is worth mentioning here that the photoinduced SmA-Cr transition has been observed in supramolecular complexes formed by interacting 4-n-alkoxy-benzoic acids with the bifunctional acceptor 4,4′-bipyridine via hydrogen bonding (Chart 5.4B) [22, 79]. Thus, a transition from the lessordered structure to highordered state occurs upon photolysis, which is, in fact, the reverse scenario when compared with other generally observed photoinduced transitions where photolysis causes a reduction in the ordering of the system. We shall describe a special case of photo-driven ordering between LC phases in a later section.
Figure 5.7 Light-stimulated interconversion of trans (EE) isomer to the cis (EZ and ZE) isomers of 1,4-diphenylbutadiene smectic LC BC-12. The SmA phase enriched with rod-like EE isomers, upon irradiation, transforms into isotropic liquid state due to formation of bent-shape EZ and ZE isomers [ref. 134].
More recently, chiral LC 2 (Chart 5.4C) derived from this 1,4-diphenylbutadiene chromophore has been confirmed to show photoinduced isothermal phase transition from layered structure, SmA phase, to N* and BP depending upon the extent of photoisomerization [135]. The focal-conic fan texture of SmA phase illuminated at 13°C below the clearing point TI with 360 nm light for 200 s, transforms into a stable oily streak texture of the N* phase. Under further irradiation, the latter pattern remains unaltered suggesting the attainment of the photostationary state; HPLC analysis shows the presence of 6% of cis isomers. Whereas, upon irradiation of the SmA phase at TI − 7°C for 100 s yields the BP with striking platelet texture, a feature characteristic of BPI/BPII; however, based on the observation that the selective reflection wavelength λmax shows strong time dependence, the phase is considered as BPI. If the SmA phase formed during the heating cycle is exposed to 360 nm light for 100 s, the BP appears with the selective reflection band at 510 nm; in fact, blue shift of the λmax occurs if the phase is continuously irradiated. The occurrence of BPI in the photoirradiated films of SmA phase has been attributed to the presence of higher amounts of cis isomers. This is quite remarkable given the fact that photoinduced occurrence of the BP had not been reported earlier in any single component LCs. However, it may be noted here that photoinduced generation of BPs/conversion from one BP to another BP/transition from BP to isotropic liquid state has been observed in some mixtures consisting of different photoactive nematic hosts and photoinsensitive chiral components [67]. For example, irradiating the N* phase of one of the mixtures with 365 nm light at room temperature, BPII is formed while heating and BPI and BPII were stabilized during the cooling cycle; this study is the first to illustrate the generation of BPs by the action of light. In another mixture, the photoinduced transitions from one BP to another have been observed; that is, when BPI is exposed to UV light for 2, 3, and 4 min, transitions to BPII, BPIII, and the isotropic phase occurs, respectively. In fact, modulation of the BP selective reflection with low levels of UV and visible light has also been established [67] in this study. The observed phenomena have been ascribed to changes in refractive indices as well as that of the helical pitch. In this context, it is worth mentioning that a photoinduced deformation in the BP structure has been demonstrated in chiral LC mixtures consisting of chiral nematic LC, chiral dopant (S811), and azobenzene compound namely, 4-dodecyloxy-4′-methoxyazobenzene [136, 137]. Irradiating the BP phase with 473 nm light, deformation in the cubic unit cell of the phase occurs with the shift in the photonic band gap. Changes in the crystal structure and platelet pattern of the BP have been established using the Kossel diffraction diagram technique.
Recently, photoinduced effects on smectic Q (SmQ) phase(s) have been revealed in an atypical chiral azobenzene compound, abbreviated as (S,S)-DCAB, where the azo-linkage bridges two biphenyl rings covalently as shown in Chart 5.4D [138]. This study is noteworthy since the SmQ phase, a chiral structure having 3D ordering, is photo-modulated, a feature that can be applied to photocontrollable switching devices. This compound exhibits two SmQ phases that were assessed based on the optical textural observations and thermodynamic property studies. The low temperature SmQ (SmQL) phase upon irradiating with UV light of 365 nm transforms into high temperature SmQ (SmQH) phase; while, the SmQH phase goes to isotropic state upon illumination. Upon switching off the UV light, the reverse transitions I-SmQH and SmQH-SmQL occur. The trans–cis isomerization of (S,S)-DCAB molecules responsible for these transitions is evidenced by electronic absorption spectra obtained in solutions with THF.
A number of light-driven transitions in photoresponsive dimers [139–144, 180] have been reported in the last decade. As mentioned earlier, odd–even effect on the phase transitional behavior, originating as a result of the spacer parity variation, is a very prominent and important feature of LC dimers; that is, the rod-like and kinked conformations of the odd- and even-members, respectively, virtually determine their thermal properties [180]. Given the fact that the photoinduced transitions largely depend on the shape of the photoactive component, the odd–even dimers exhibit interesting photoinduced phase behavior. For example, several light-driven isothermal phase transitions have been observed in single component dimers D-4 to D-21 (Chart 5.5A) that are made by joining cholesterol and azobenze segments through a flexible spacer [140–143]. The SmA and N* phases formed by such dimers transform into the isotropic phase upon irradiation with 365 nm light [140]. Notably, a hypsochromic shift occurs in the selective reflection wavelength (λmax) of the equilibrium N* phase when it is exposed to UV light; the magnitude of the shift depends on the irradiation energy, temperature, and the length of alkyl tail of the azobenzene segment. These properties have been exploited in a rewritable full color image recording in photon mode [140]. The shift could be due to the isomerization-driven approach to the isotropic phase or as the authors speculate, the bent cis form has a smaller λmax. These photosensitive dimers have also been employed as dopants with a dicholesteryl ester to realize a stable glassy N* state [140]. In these materials, irradiating the SmA phase with 366 nm light, the system first transforms to the N* phase and with continued illumination changes to the isotropic phase; and thus, SmA-N* and N*-I transitions have been observed [142]. In another dimer, exhibiting SmC*, SmA, and N* phases, a direct photoinduced transition from the SmC* phase to isotropic phase occurs without passing through other two phases [143] and thus, the SmC*-I transition has been observed. The percentage of cis isomer required for SmC*-I, SmA-N*, and SmA-I transition were estimated with the help of HPLC to be 30%, 6%, and 19%, respectively. It must be mentioned here that the effect of photoisomerization on the order parameter (spontaneous polarization) and dynamics (response time) has been investigated in the SmC* [145–152], antiferroelectric SmC* (SmCA*) [153, 205], and chiral smectic I (SmI*) [154] phases on their approach to SmA and SmC* phases, respectively. These studies show that polarization diminishes and the response becomes faster upon photoisomerization.
Chart 5.5 (A) General molecular structure of chiral azobenzene-based LC dimers exhibiting various reversible photoinduced transitions [refs. [140]–143]. (B) General molecular structure of photoactive dimers derived from 1,4-diphenylbutadiene showing light-induced SmA-N* and N*-I phase transitions [ref. 144].
Photoinduced SmA-N* and N*-I transitions were seen in analogous dimers D-22 to D-24 (Chart 5.5B) comprising non-azobenzene chromophore, viz., 1,4-diphenyl-butadiene core linked to cholesterol segment through an oxyalkoxycarbonyloxy (carbonate) spacer [144]. In these compounds, modulation of the reflection color of the N* film over the entire visible range has been achieved. The color images could be stored by rapidly quenching the irradiated film to 0°C. Recently, such observations have been made with the N* phase formed by LC trimers also [155].
A photoinduced reversible SmC*-N* phase transition under the influence of an applied electric field has been reported in mixtures consisting of an achiral smectic C (SmC) liquid crystal (4-hexyloxyphenyl-4-octyloxybenzoate: HOPOOB), chiral dopant [4-(methyl-heptyloxycarbonyl)phenyl 4-hexyloxybenzoate: S811], and azobenzene derivative (4-diethyl-aminoazobenzene) as a dopant in 3–7 wt% concentration (Chart 5.6) [156]. The SmC* phase formed upon cooling the N* phase held under the influence of a DC electric field shows uniform unidirectional layer alignment in the entire region of the rubbed cell. Upon reversing the polarity of the field, molecules tilt with respect to rubbing direction; further, the photolysis of this state gives rise to N* phase where the molecules tend to orient along the rubbing direction and in the absence of UV light, the N* phase transforms into SmC* phase. Thus, the photoassisted control of smectic layer alignment/switching, which appears to hold promises in fabricating display, optical memory, and optical grating devices, is achieved.
Chart 5.6 Molecular structures of a liquid crystal (HOPOOB), chiral dopant (S811), and azo dye used to demonstrate reversible SmC*-N* phase transition under the influence of an applied electric field [ref. 156].
5.2.7 Photoinduced Transitions Involving Columnar Phase
From the foregoing discussion, it is quite evident that a variety of PIPT have been established using different photoresponsive LC motifs mostly of rod-like shape (calamitics). However, it is quite surprising that there is hardly any such study on disk-like (discotic) molecules [157], despite the fact that a number of photoactive discotics have been designed and synthesized [158–161]. Very recently, Norikane and coworkers [162] reported photoassisted transition from the Col to the isotropic phase in a macrocyclic compound 3b comprising three fused-azobenzene mesogenic segments (Figure 5.8). Upon photolysis, the room temperature Col phase of the compound transforms into isotropic liquid where nonplanar (bent) molecules formed due to their E–Z isomerism destabilize the stacking of the molecules in columns; this process is reversible due to thermal back relaxation and quite sensitive to even small fractions of Z-isomer.
Figure 5.8 Schematic representation of self-assembly of photoactive discotic 3b into columnar LC structure which upon photolysis transforms into isotropic phase due to trans–cis isomerism where the flat disks fold-up leading to the destabilization of columnar organization. Note that when the thermal relaxation from cis to trans isomers occurs, the isotropic liquid state transforms into the Col phase [ref. 162].

5.3 Detailed Account of PIPT in Specific Systems
In the following sections, we will look at the influence of certain parameters on the light-driven transitions. Specifically, we describe how parameters such as surface fields, elevated pressure, anisotropy reducing component, magnitude of orientational order, and electric field have a bearing on the photoinduced N-I transition. Also elaborated are the phenomena of dynamic self-assembly of a layered phase, tilt susceptibility of chiral smectic system, and primary and secondary photo ferroelectric effects in an antiferroelectric LC. Finally, a description of PIPT in systems composed of bent-core molecules is given.
5.3.1 Surface Fields
The importance of surface forces in controlling the dynamics is well known. For example, the Freedericksz transition with a well determined threshold requires proper surface forces and the relaxation to the equilibrium conditions is governed by the nature of the surface. The influence of such surface forces on PIPT, especially its dynamics, has been studied by confining the liquid crystalline material in networks. The employed networks were of two different types: a rigid polymer matrix and a fragile network of hydrogen-bonded aerosil particles. This section describes the results of these measurements wherein the sample is subjected to a restricted geometry.
5.3.1.1 Aerosil-LC
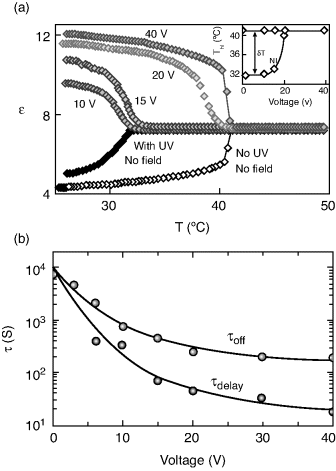
A convenient way of creating a confined geometry is to disperse aerosil particles in a liquid crystal medium [164]. These silica particles of ~7 nm diameter and capped with hydrophilic or hydrophobic agents, form in the presence of a fluid medium—such as nematic—a fragile network of hydrogen bonds. The concentration or density as it is termed in these systems, of the aerosil particles, is given by ρa = ma/VLC, where ma is the mass of aerosil and VLC is the volume of LC material. ρa controls the strength of such a network, which could span from the soft to the rigid gel regime. The voids (the regions enclosing the LC molecules) define the scale of confinement, and thus ρa becomes the control parameter of the restricted geometry. Unlike the standard cell in which the surface–LC interactions arise only at the two bounding plates, the aerosil LC system results in such interactions in the “bulk” of the material also and therefore serves to enhance the influence on the static and kinetic characters of PIPT. LCM3, used as the host substance in the results to be described below consisted of 3.3 wt% of EPH in 4-n-heptyl-cyanobiphenyl (7CB). Figure 5.9 shows the differential scanning calorimeter (DSC) scans [167] obtained across the I-N transition for LCM3 and a soft gel composite (ρa = 0.03 g/cm3) at a cooling rate of 1°C/min. While LCM3 exhibits a single peak, the aerosil mixture shows a twin-peak profile, more clearly seen at a cooling rate of 0.3°C/min (see inset of Figure 5.9). The observation of such a two-peak profile in an aerosol–LC composite has been associated [163, 167] with the development of nematic order from the isotropic phase through a two-step process. It is caused [165] due to a crossover from a random-dilution regime, where the silica gel couples to the scalar part of the nematic order parameter, to a random-field regime (occurring at a lower temperature), in which the coupling induces distortions in the director field. It should also be noted that the high temperature peak is much sharper compared to the low temperature peak. Since the high temperature peak is due to the appearance of the nematic domains in the isotropic phase, having a weak coupling to the aerosil network, its thermal signature is weaker than the low temperature peak. Upon isomerization of the photoactive component, the signature of this two-step process becomes prominent in the thermal and temporal variation of the dielectric constant of the sample, and also the temperature difference between the two steps increases. Surprisingly, just like TNI, ΔTNI, the photoinduced shift in TNI, also varies non-monotonically with the aerosil density (see Figure 5.10). These results favor the random-field model in which the quenched LC is simply distributed randomly in space [166, 167]. The gel network also affects the kinetics of the photoinduced as well as the back relaxation process. The associated time scale again has a non-monotonic dependence on the aerosol density (Figure 5.11).
Figure 5.9 DSC scans at a rate of 1 K/min in the bulk and aerosil composites (aerosil concentration, ρa = 0.03 g/cm3) of 7CB + 3.3% EPH LC mixture, in the vicinity of the N-I transition. Notice the single peak of the bulk material gets split into two for the aerosil composite, a feature that is clearer when the cooling rate is reduced to 0.3 K/min, as shown in the inset [ref. 167].
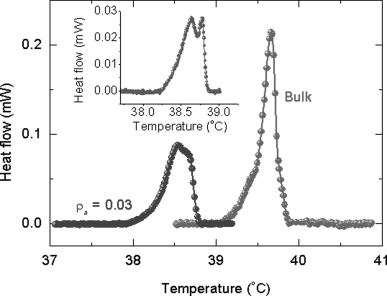
Figure 5.10 Influence of the aerosil composition on the transition temperature TNI and the UV-induced shift in the transition temperature ΔTNIUV. The inset shows that the ratio of the two parameters is only weakly dependent on the aerosil concentration. The line drawn through the data points is a fit to a straight line and the error bars indicate 95% confidence limit [ref. 167].
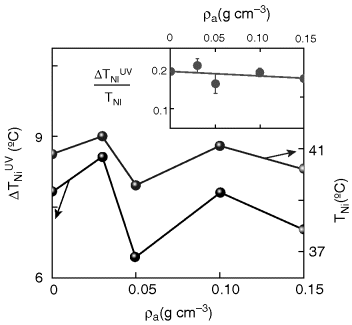
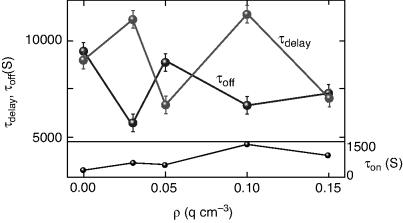
Figure 5.11 Dependence of the τdelay and τoff, the delay in the initial response and the total time taken to complete the back relaxation, respectively when the UV is switched off, on the aerosil composition. The response time τon for the photoinduced transition is shown in the bottom panel. Notice that all the responses vary non-monotonically with the aerosil composition [ref. 167].
5.3.1.2 Polymer Matrix
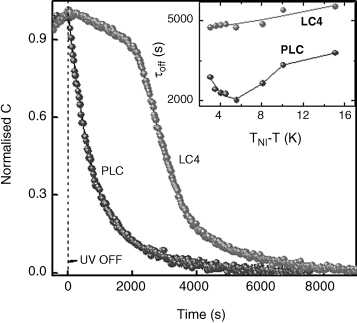
In comparison to the aerosil-LC case discussed above, LC sample dispersed in a polymer matrix (referred to as a polymer dispersed liquid crystal-PDLC system) forms a different kind of confinement. Unlike in the case of the aerosil-LC, the confining matrix of the polymer is more rigid and therefore the LC–surface interactions, which are again present not only at the bounding plates but also at each LC–polymer interface, should be stronger. The PDLC sample employed was composed of a photocurable prepolymer (Norland NOA65) and a LC mixture (Merck-E7 + 5% EPH, referred to here as LC4) in the ratio of 62:38; we refer to the polymerized sample as PLC. Figure 5.12 shows the TBR process for the LC4 and PLC samples [168]. The main difference in the behavior of the two samples was the substantial time delay (~2000 s) between the time of switch off, and the instant at which there is an abrupt decrease in sample response marking the onset of the I-N transition, typical of unconfined samples (including the LC4 system), but absent for the PLC sample. However, after the abrupt variation further change in the response occurs at the same rate in both the cases. This behavior can be explained by applying the fact [169] that nematic order is known to persist at the surface even after the bulk has transformed into the isotropic phase. When UV is switched off this surface nematic order would promote the return of the photoinduced isotropic to the nematic phase in the bulk. While for the LC4 sample the surface order is present only at the two substrate surfaces, for the polymer-based samples the presence of the polymer matrix provides “virtual surfaces” even in the bulk, at each LC–polymer interface, causing an acceleration of TBR and reduction of the delay time. Once the bulk starts recovering the N phase, the effect of the surface diminishes and therefore after the delay period the behavior of the two samples is quite similar. Owing to these features the total TBR time is twice as fast for the PLC sample in comparison to that for the LC4 (see inset of Figure 5.12).
Figure 5.12 The thermal back relaxation processes for LC4 and PLC samples probed as a function of the normalized sample capacitance C. After the UV is switched off, the LC4 sample responds slowly and exhibits a substantial time delay between the time at which the UV is switched off and the instant at which there is an abrupt decrease in C, marking the onset of the I-N transition. In contrast the PLC sample, with the polymer matrix, responds instantaneously and has no time delay. The inset shows the dependence of τoff as a function of reduced temperature for the two samples. Reprinted with permission from ref. 168, Copyright 2003, American Institute of Physics.
5.3.2 Influence of Anisotropy Reducing Component
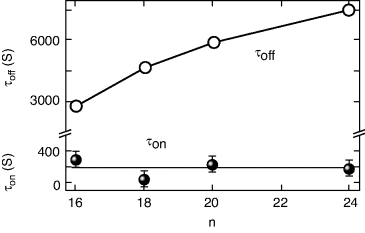
Octadecane and other such similar long chain alkanes have only flexible units and thus per se are not favored to support the formation of liquid crystalline phases. Even when added to materials that already exhibit liquid crystalline phases they try to destabilize the liquid crystallinity [170]; at low concentrations (as it was the case in our experiments), the small biphasic region suggests that the structural incompatibility between the LC system and the alkane is still tolerable on a macroscopic scale. In the isotropic phase, due to the preference of the alkane molecules to exist in the coil-form, there would be less difference between the local order in the vicinity of the alkane and that of the entire medium. On the other hand, in the N phase the alkane molecules are forced to deviate from the coil-form to be compatible with the environment. But given an opportunity to deviate from this situation, the alkane molecules may hinder the liquid crystalline molecules in the vicinity from achieving nematic order. For the temperature-dependent transition, such an influence will lower the transition temperature, a feature generally observed for liquid crystalline mixtures containing alkanes. In the case of photoinduced transition, the process is an isothermal one. Thus a possibility is that in the absence of any isomerization-driven torques (e.g., presence of radiation to facilitate reverse isomerization) the local disorder of the alkane molecules will retard the relaxation of the system from the photoinduced isotropic phase, thereby increasing TBR by an order of magnitude [171]. Further, the length of the alkane employed also has a significant effect: a factor of 8 increase in TBR is seen when n, the number of carbon atoms in the alkane, is increased from 16 to 24 [172]; in contrast, the time duration for the photo-driven N-I process is hardly affected (see Figure 5.13).
Figure 5.13 Influence of the length of the alkane, characterized by n, the number of carbon atoms in its chain, on the response times for the UV-on (τon) and UV-off (τoff) processes at a fixed relative temperature of 20°C with respect to the clearing point. The ternary composite employed has the primary mixture consisting of hexyloxy cyanobiphenyl doped with 5 wt% EPH, to which the alkane is added at a concentration of 2.5 wt%. The lines are merely guide to the eye [ref. 171].
5.3.3 Influence of the Orientational Order
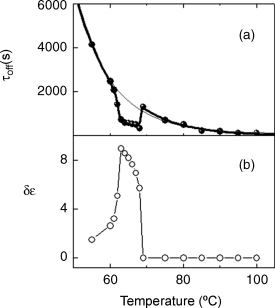
The orientational order of the nematic phase can be expected to govern the kinetics of the thermal back relaxation. However, contrary to expectations, τoff exhibits [173] a non-monotonic variation with temperature having a pronounced reduction in the finite ΔTNI region bracketed by the two transition temperature without (Tno-UV) and with UV (Twith-UV). Treating δε, the difference in the dielectric anisotropy εa (=ε![]() − ε⊥) between the equilibrium and photo-stimulated situations, as a good measure of the orientational order a simple correlation between τoff and δε has been found (see Figure 5.14):
− ε⊥) between the equilibrium and photo-stimulated situations, as a good measure of the orientational order a simple correlation between τoff and δε has been found (see Figure 5.14):
Figure 5.14 Thermal variation of the response time (a) τoff and (b) δε, the difference in the dielectric anisotropies for the no-UV and with-UV. The thick line in (a) is a fit to Eq. (5.1) and the thin line represents Arrhenius behavior [ref. 173].
5.3.4 Acceleration of TBR by an External Field
As discussed in previous sections, TBR, marking the recovery of the state existing before the photo-stimulation, is a slow process, extending from a few tens of minutes to days. Illuminating the sample with another radiation, typically in the blue region of the visible spectrum, is an established method of reducing this time scale. Now, we describe a recent finding [174] wherein an applied electric field can drastically alter the photoinduced thermal behavior and also accelerate the return to the equilibrium state. The fixed frequency dielectric constant data obtained for a planar sample in the absence of an electric field (except for the probing field) and when DC voltages of different magnitude are applied to a material with a positive εa are shown in Figure 5.15a. It should be noted that owing to +εa, the applied voltage, above a Freedericksz threshold (2 V for the sample used) causes a reorientation of the molecules from the planar to the homeotropic configuration. As the voltage is increased, ΔTNI, the UV-induced shift in the transition temperature, is diminished to the extent that above a certain voltage there is no shift at all [174], resulting in an interesting temperature–voltage phase diagram (inset of Figure 5.15a). The striking influence of the field on TBR is shown in Figure 5.15b: τdelay reduces by two orders of magnitude and τoff by a factor of about 20 [174]. In a later section, we shall show that the observed behavior is universal and applicable to transitions involving different LC phases and discuss the possible reasons.
Figure 5.15 (a) Influence of the electric field (with the voltages indicated against each data set) on the dielectric constant behavior across the N-I transition in the presence of the UV radiation (filled circles). For comparison, temperature dependence of the dielectric constant ε obtained without the UV radiation (open symbol) is also plotted. Due to the positive dielectric anisotropy of the sample the application of the electric field reorients the molecules from the surface determined planar to the high-field driven homeotropic orientation. As a consequence ε decreases across the N-I transition unlike in the field-off case. For the sake of convenience of presentation, the data in the isotropic phase has been matched between different sets. Inset: Temperature–voltage phase diagram obtained without (open circle) and with UV radiation (filled circle). Notice that there is no UV-induced diminution in the transition temperature for voltages > 20 V. (b) Voltage dependence of the two time scales τdelay ![]() and τoff
and τoff ![]() associated with the relaxation process, with the lines describing fit to an exponential function [ref. 174].
associated with the relaxation process, with the lines describing fit to an exponential function [ref. 174].
5.3.5 Effect of Elevated Pressure
Pressure being a thermodynamic variable, it can be expected that subjecting the sample to elevated pressures influences PIPT. Such studies demonstrated [175–177] the feature that application of pressure reduces ΔTNI, even leading to complete absence of such a shift above a certain pressure. It was also found that Po, the pressure at which such suppression takes place, can be tuned by IUV, the magnitude of the actinic light intensity: Po increases as the intensity is increased. Further, both ΔT and Po vary strongly with IUV at low intensities but have a limiting type of behavior for higher values of IUV. This trend is qualitatively similar to the variation of ΔTNI with IUV at room pressure. The behavior of Po is also similar to that of the dependence of the E–Z isomer conversion efficiency on IUV. Pressure also strongly alters the kinetics of the photoinduced transition and its reversal. For quantitative description of the observation, we define four periods τ1 and τ3 as the delay times, and τ2 and τ4 as the response times associated with the UV light being switched on and subsequently off. With increasing pressure, while the UV-on times, τ1 and τ2, show a moderate increase, τ3 and τ4 connected with the recovery of the equilibrium phase, exhibit an exponential decrease.
A standard nematic host (8OCB) doped with n = 4–11 homologues of a dimeric azobenze series [α,ω-bis(4′-n-butylazobenzene-4-oxy) alkane, BBAOA for short] as guests, has shown that the parity and length of the dimers affects the static and kinetic characters of PIPT [177]. For example, not only the transition temperature TNI but the shift ΔTNI, also exhibits an alternation with the parity of the spacer in the dimer molecule. Figure 5.16a and b depicts the pressure dependence of the N-I phase boundary in the absence of, and upon UV illumination for the M4 and M5 mixtures (the numeral indicates the number of methylene units in the spacer of the photoactive compound). Although ΔTNI diminishes with increasing pressure and finally vanishes for both the mixtures, Po is substantially different. Indeed its value alternates with the parity of the spacer in the dimer, with the sign of alternation opposite to that of ΔTNI (see Figure 5.16c).
Figure 5.16 Pressure–temperature phase diagrams showing the N-I phase boundary obtained in the absence of (red symbols, without) and with UV radiation (blue symbols, with), for the representative mixtures M4 (a) and M5 (b), respectively. The photoinduced shift ΔTNI decreases with increasing pressure becoming zero at a pressure of Po. (c) Odd–even dependence of ΔTNI and Po on the spacer length of the dopant dimeric molecule. (d) Dependence of the total time (delay + response time) for the UV-on (tso) and UV-off (tsf) processes on the applied pressure at a constant reduced temperature of Tred = 5°C for the mixture M4 [ref. 177].
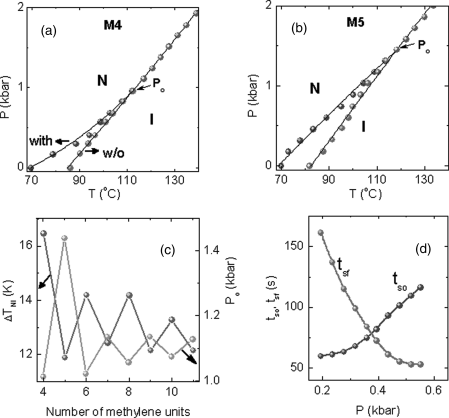
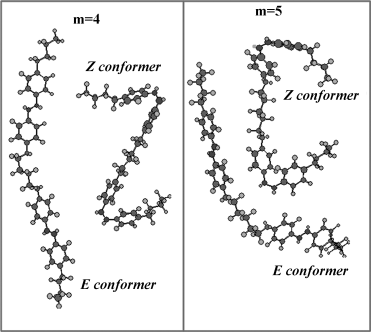
Now, let us look at the possible reasons for the features observed in the high-pressure experiments. Isothermal measurements [178] suggest a decrease in the transition volume with increasing pressure. Consequently, with pressure there should be a reduction of the intermolecular space available for the azobenzene molecule to take a bent shape, which manifests as an opposition by the system for the formation of the Z isomers. As a result, the magnitude of ΔTNI, dependent on the concentration of the Z isomers also becomes smaller as the pressure is increased and finally vanishes. This effect can be overcome, at least to a certain extent, by increasing IUV thereby forcing the E–Z isomerization, leading to at least a partial restoration of the photoinduced shift in the transition temperature [176]; the balance between these two opposing forces decides the value of Po for a certain IUV. This fine balance between the opposing influences of the energy pumped and the pressure on the photoisomerization, can also explain the pressure dependence of the kinetics; the total (delay + response) switch off duration tsf decreases with increasing pressure, whereas the total switch on duration tso increases moderately (see Figure 5.16d). The odd–even effect on ΔTNI caused by the parity of the spacer can be understood as follows. The E conformers of the even and odd spacers have respectively a somewhat elongated shape and a significantly bent shape (see Figure 5.17). Upon isomerization, whereas the even members assume a “Z” shape, the odd members take a “C” shape. This difference in the shape causes the projected length of the photoactive dopant to exhibit an alternation with n, the magnitude of the alternation being much larger for the E conformers than for the Z conformers [179, 180]. The larger change in the dimension of the even member dimers results in a larger value of ΔTNI corresponding to their odd counterparts.
Figure 5.17 Minimum energy configurations in the E and Z conformer states for representative even (m = 4) and odd (m = 5) spacer dimer molecules.
In the previous sections, we considered the influence of photoisomerization on the N-I transition. Next we consider achiral as well as chiral systems possessing smectic phase(s). Generally, a phase transition caused by the E–Z isomerization results in a reduction of the ordering of the medium, i.e., the transformation is an order to disorder transition. Further, as a rule, the photoinduced phase would in any case occur in the thermal cycle. Exceptions to these features were found in the recent experimental results, as discussed in the following.
5.3.6 Dynamic Self-Assembly of the Smectic Phase
The phenomenon of reentrance has attracted a great deal of attention in a variety of condensed matter systems such as multicomponent systems, superconductors, solid ferroelectrics, and liquid crystals. A phase is said to be reentrant if it occurs both above and below another phase when a thermodynamic field like, e.g., temperature or pressure is varied. Perhaps the earliest example is of the nicotine system investigated by Hudson [181, 182]. In liquid crystals, the first such example was reported in a binary phase diagram at atmospheric pressure [183] later for a single component material at high pressures [184], and subsequently at room pressure [185, 186]. Since then a number of single-/multi-component materials have been reported to exhibit the reentrant sequence, either at atmospheric or at elevated pressures. The studies to be described below were on the reentrant nematic phase seen in a temperature–concentration binary phase diagram: specifically, for certain compositions, the system containing a photoactive guest component exhibits a nematic−smectic A−reentrant nematic (N−SmA−Nre) phase sequence on cooling from the isotropic phase. Upon UV illumination, the SmA and the high temperature N phases transform into N and isotropic phases respectively, as indeed expected, since photoisomerization can result in the “melting” of the phase, or in other words, transformation to a lessordered state. The Nre phase, however, transforms to a more ordered state, viz., the SmA phase [187]. Even more exotic behavior was observed [188, 189] for a mixture which did not exhibit the SmA phase in the absence of UV radiation. The self-assembly to the layered phase occurred with the UV radiation acting as a stimulant and existed only as long as the radiation was present.
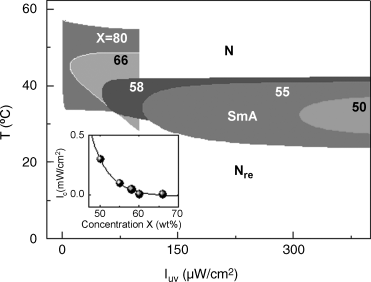
The self-assembly of the layer structure in the above two cases (of dynamic character in the second one) has been explained using a combination of the photo-driven nanophase segregation mechanism [190] and the frustrated spin-gas model [191], the essentials of which are given in the following. In the equilibrium situation (absence of UV radiation) the photoactive molecules, in their rod-like E form are easily accommodated into the smectic layers. In contrast, the photoinduced Z isomers having a bent form not compatible with the shape of the host molecules are expelled to a region between the host molecule layers creating nanophase segregation. The frustrated spin-gas model [191] was essentially proposed to explain the reentrant behavior in materials, such as the host compound in the above-mentioned studies [188, 189] in which the molecules have a strong terminal polar group. Frustration is introduced by considering the possible terminal dipole configurations of a triangular lattice of the molecules. Relative stability of the SmA or the N (Nre) phase is compared by looking at the contributions of the dipole triplet to the free energy. In such a scenario, in the absence of the photoisomerization-driven segregation the molecules are free to liberate along the director direction, which promote the N or the Nre phase. The nanophase segregation due to photoisomerization will, in the segregated layers of the photoactive Z isomers in their bent form, create an alien atmosphere for the host rod-like molecules, thus disfavoring libration. Consequentially a layered arrangement as in the SmA phase is favored, as indeed established by X-ray (Figure 5.18) and magnetic field driven Freedericksz transition experiments (Figure 5.19). The texture changes seen under a polarizing microscope when the material transforms from the equilibrium Nre to the photoinduced SmA phase are shown in the inset of Figure 5.18, providing a clear support for the photo-driven layered structure. The photostabilization of the SmA phase allows one to map novel temperature–UV intensity (T–IUV) phase diagrams, such as the one shown in Figure 5.20, with the critical intensity (Ic) needed to photoinduce the SmA increasing drastically as the concentration of the non-smectic component in binary non-photoactive host decreases (see inset of Figure 5.20). It may be pointed out that the UV intensity, used as a variable here, is a non-thermodynamic quantity, but mimics, in a limited sense, the role of a thermodynamic parameter like, for example, pressure. Also, it is worth noting here that these phase diagrams are obtained under non-equilibrium conditions as the existence of the SmA phase is dynamically driven.
Figure 5.18 X-ray diffraction profiles of the (a) equilibrium Nre and (b) photoinduced SmA phases. As expected the Nre phase shows a broad and diffuse peak whereas the SmA has an intense and sharp profile. The lines are fits to a Lorentzian expression. The inset shows the corresponding polarizing microphotographs in the two phases: (a) Nre and (b) SmA [ref. 188].

Figure 5.19 The magnetic field dependence of the sample dielectric constant in the photoinduced SmA (curve B) to the equilibrium Nre (curve A) phases. The time lapse profiles C–F show the behavior during the thermal back relaxation of the system after the UV beam is switched off. The temporal variation of the critical Freedericksz threshold field Hc during this relaxation is shown in the inset [188].

Figure 5.20 Partial temperature–UV intensity (IUV) phase diagrams for various concentrations (X in wt%) of the non-smectic component in the host (non-photoactive) mixture, but with a fixed (4 wt%) concentration of the UV-active compound, to demonstrate the photostabilization of the SmA phase in systems (X = 50–66) lacking it in the absence of UV light; the filled area represents the thermal range of the SmA phase, whereas N and Nre phases exist above and below it respectively. For comparison, the diagram for X = 80 is also shown for which the SmA exists in the thermal cycle and its range increases with UV intensity. Inset: As X decreases the critical UV intensity (Ic) needed to induce the SmA phase increases drastically [ref. 189].
As stated earlier, an applied electric field has been reported to accelerate the relaxation of the Z isomers to their E form, and consequent recovery of the equilibrium phase. The effect of the electric field is not specific to the N phase [174] or even to LC materials [192]. Here we quote an example of the reentrant system. Unique temperature–electric field phase diagrams of a liquid crystal exhibiting isotropic–nematic–smectic A–reentrant nematic sequence, mapped using light transmission as probe reveal that the electric field influences all the transitions, but the effect is maximum on the equilibrium reentrant nematic to the photoinduced smectic A transition [193]. Temporal measurements performed under non-equilibrium conditions to study the dynamics of both the photochemical and the back relaxation processes across the different transitions, show that the electric field is indeed observed to accelerate the thermal back relaxation in each case; especially the recovery of the reentrant phase is hastened by three orders of magnitude in time. A schematic diagram representing the molecular arrangement for the photo-driven process across the three transitions, N-I, SmA-N, and Nre-SmA is given in Figure 5.21.
Figure 5.21 Schematic representation of the molecular arrangements in the equilibrium (left panels) and photo-driven (right panels) states. Notice especially the nanophase segregation in the photo-stimulated SmA phase (panel c). The cyan objects stand for the host non-photoactive molecules with a cyano termination. The red-straight objects are the azobenzene (EPH) molecules in their E form, which upon isomerization assume a bent form (bent red objects). The left pointing arrows emphasize the fact that the application of an electric field accelerates the return of the equilibrium phase owing to a swift back relaxation of the Z isomers to their E form, which otherwise is a very slow process.
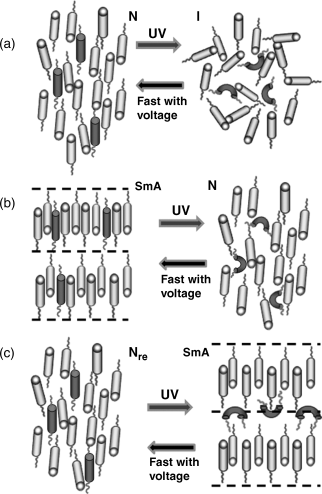
Now we look at the possible causes for the observed acceleration. While simple dielectric or flexoelectric coupling can be ruled out as the possible cause for the electric field-determined acceleration of the recovery of the E isomers, the electrode polarization as the reason may still be operative in the N-I as well as the reentrant cases. However, the different magnitudes of acceleration for the different transitions in the same system, and also that the values are non-monotonical with temperature indicates that there could be other influences also. It is perhaps that the electric field directly influences the azobenzene molecule itself with the host molecules hardly playing any role.
A theoretical model that is under consideration in this regard is the density-functional-theory-based calculation [194], which proposes the possibility to induce the isomerization of azobenzene in the gas phase by an electric field; this model has been used to explain electric field-induced isomerization of an azobenzene derivative on the gold surface by a scanning tunnelling microscope (STM) tip [195]. The theory considers that in the presence of an electric field, the potential-energy surface related to a reaction path can be deformed, thus leading to an effective lowering of the isomerization barrier. Such an effect is argued to depend on the orientation of an intrinsic dipole moment (if present) and also on the polarizability of the molecule. The essential point is that application of an electric field reduces the potential barrier between the E and Z isomers and therefore favors a quicker return of the photoactive molecule to the equilibrium form. Qualitatively this theory can explain the influence of the electric field on the static as well as dynamic properties that have been observed in our experiments. A point that should however be emphasized is that the actual electric field in our experiments is (even at the highest voltage of 20 V) three orders of magnitude smaller than in the STM experiments. It is possible that the anisotropic ordering of the LC environment lower the actual field required.
5.3.7 Tilt Susceptibility Behavior Across SmA-SmCα* Transition
The SmC phase is the tilted analogue of the SmA phase having the director at an angle to the layer normal direction. Investigations on this phase, especially when composed of chiral molecules (SmC*) have attracted much attention since the phase displays ferroelectric properties. A variant of the SmC* phase, called as the antiferroelectric SmC* or SmCA* phase, was reported in 1989 in which the molecules in neighboring layers are tilted from the smectic layer normal in almost opposite directions [196]. Subsequently, other sub-phases like the ferrielectric SmCγ* and SmCα* phases have been discovered [for reviews on the antiferroelectric smectic and sub-phases see 197, 198]. Resonant X-ray scattering experiments [199] have established that the SmCα* phase has a short-pitched incommensurate helical structure extending over a few layers. Investigations on the effect of light in a system exhibiting the SmA-SmCα* phase transition using dielectric spectroscopy show that at any given reduced temperature, T − Tc (Tc is the SmA-SmCα* transition temperature) the UV light enhances the magnitude of the tilt fluctuations [200, 201]. This is the opposite of the effect induced by ordering fields such as electric or magnetic fields [202]. Figure 5.22 presents the dielectric absorption spectra at a fixed temperature after the UV radiation is turned on: with elapsing time, the soft mode relaxation frequency fs connected with softening elastic opposition to tilt fluctuations increases while the strength of the mode εa decreases before reaching a photostationary state in about a few minutes. The detailed temporal behavior of these two parameters brings out the influence of the magnitude of the UV intensity (see Figure 5.23). The variation of fs with time is not the same at all UV intensities, with two different regions demarked by a critical intensity Icr. For IUV < Icr (region I), fs decreases monotonically with elapsed time. In region II, with IUV > Icr, fs diminishes immediately after the UV is on, but reverses its trend after a certain time interval; the time at which the reversal is seen decreases for higher intensities. Concomitant changes are seen in the dielectric strength. Comparing this behavior with the well-known thermal variation of these parameters [201], such a reversal in the trend signifies the photoinduced and isothermal SmA-SmCα* transition. Owing to the striking similarity between the thermal and IUV dependence of the relaxation parameters, the latter data have been analyzed in terms of the equations of a Landau model for the thermal SmA-SmC* transition, expressing fs as
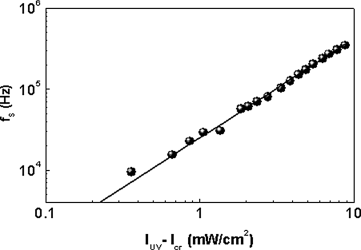
Here, ![]() represents the amplitudes above and below Icr, the critical intensity to induce the isothermal transition which plays the role of transition temperature for the case of the thermal transition. In the thermal case, the exponent γ describes the critical behavior of the susceptibility near a critical point, having values dependent on the universality class to which the transition belongs: 1.24 for the 3D Ising and 1.316 for the 3D XY. The fitting to Eq. (5.2), shown as solid line in Figure 5.24 yields γ = 1.23 ± 0.04, which within error bar is in agreement with the theoretically prediction for the Ising model [203]. It is worthwhile to recall that the specific heat data analyzed using correction-to-scaling terms seemed to favor the XY model, although description with the Ising model could not be unambiguously precluded [204]. In our opinion, the third possibility of the measured exponent being an effective one due to the crossover from mean field (γ = 1) to 3D XY (γ = 1.316) regimes, cannot also be ruled out. A caveat that should be added concerning the photo-driven transition data is that there is no a priori rule to say that the photoinduced isothermal transition should behave the same way as a thermal transition.
represents the amplitudes above and below Icr, the critical intensity to induce the isothermal transition which plays the role of transition temperature for the case of the thermal transition. In the thermal case, the exponent γ describes the critical behavior of the susceptibility near a critical point, having values dependent on the universality class to which the transition belongs: 1.24 for the 3D Ising and 1.316 for the 3D XY. The fitting to Eq. (5.2), shown as solid line in Figure 5.24 yields γ = 1.23 ± 0.04, which within error bar is in agreement with the theoretically prediction for the Ising model [203]. It is worthwhile to recall that the specific heat data analyzed using correction-to-scaling terms seemed to favor the XY model, although description with the Ising model could not be unambiguously precluded [204]. In our opinion, the third possibility of the measured exponent being an effective one due to the crossover from mean field (γ = 1) to 3D XY (γ = 1.316) regimes, cannot also be ruled out. A caveat that should be added concerning the photo-driven transition data is that there is no a priori rule to say that the photoinduced isothermal transition should behave the same way as a thermal transition.
Figure 5.22 Dielectric loss spectra taken in the SmCa* phase (1°C below the transition, obtained before t = 0) and at elapsed times (indicated against each curve, in seconds) after the UV illumination is turned on. Notice that on UV illumination, initially the relaxation frequency (~peak frequency of the profiles) shifts to a lower value (t = 10 s) scan and then to higher values, reaching a limiting value for longer time scales [ref. 201].
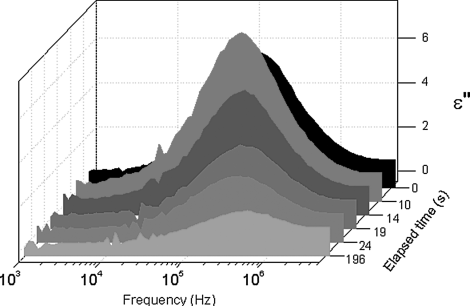
Figure 5.23 Evolution of the soft mode relaxation frequency (fs) in the SmCa* phase as a function of time for UV-on and subsequent UV-off situations. The number mentioned above each dataset indicates the intensity of the UV light (in mW/cm2) employed. For the sake of clarity the data sets have been shifted by arbitrary amounts in the vertical direction. It may be noted that before UV-on the fs values coincide for all data sets [ref. 201].
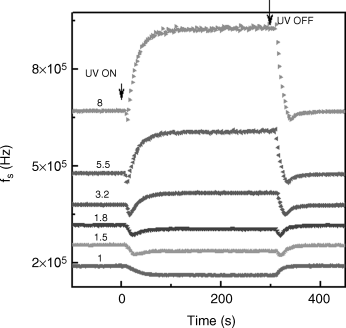
Figure 5.24 Double logarithmic plot of the UV intensity dependence of the saturated values of the relaxation frequency, with the slope of the straight line (a fit to Eq. (5.2)) yielding γ = 1.23 ± 0.04 and a critical intensity Ic value of 1.2 ± 0.1 mW/cm2 [ref. 201].
5.3.7.1 Polarization-Tilt Coupling in an Antiferroelectric System
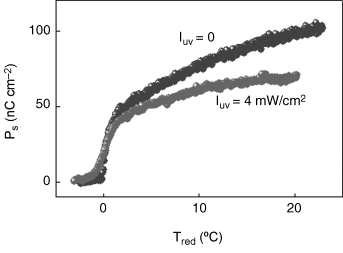
The better known of the smectic C antiphases, is the antiferroelectric smectic C (SmCA*) phase [196]. In this phase, the molecules in the neighboring layers are tilted from the smectic layer normal in almost opposite directions; in contrast the tilt is synclinic in the SmC* phase. Generally, the SmC* phase intervenes between the SmA and SmCA* phases. But in certain materials there can be a direct transition from the Sm-A to the SmCA* phase. A detailed study on the effect of photoisomerization in a binary mixture (referred to as AFM hereafter) composed of TFMHPOBC and 5% by weight of the guest UV-active dopant EPH has been reported [205]; this system exhibits a direct transition from the SmA to the SmCA* phase. The two-peak sample current response to an applied triangular wave characteristic of the SmCA* phase is employed to determine the electric polarization (Ps) as a function of temperature in the absence of and upon illuminating the sample with IUV = 4 mW/cm2. The gross trend of the temperature dependence of Ps remains the same with and without UV. However, two distinguishing differences result from photoisomerization: () there is a reduction in Tc (the SmA-SmCA* transition temperature) and () the saturated value of Ps seen well below Tc is substantially lower than without UV. The first feature, routinely seen for PIPT as discussed above, is labeled as secondary photoferroelectric effect [150]. Even after taking care of the reduction in the transition temperature, a large (30%) diminution in Ps found experimentally (Figure 5.25) should be accounted for. We suggest that this may be associated with a change in the polar ordering and/or the transverse molecular dipole moment and refer to it as primary photoferroelectric effect (PPE), and in the following look for possible causes. In ferroelectric as well as antiferroelectric LC systems the tilt angle of the molecules (θ) with respect to the layer normal is the primary order parameter and the electric polarization Ps is a secondary order parameter. Owing to a strong coupling between these two parameters, the primary photoclinic (light-driven tilt of the molecules) effect, although can be expected to be the cause for PPE, is not borne out by the experiments. The nanophase segregation mechanism, described above, is a possibility, but ruled out however, owing to the fact that upon UV illumination the maximum observed increase in layer spacing, and the consequent change in the dipole moment/unit volume is too small to account for the measured decrease in Ps. We suggested [205] that the cause lies in photoisomerization altering the coupling between Ps and θ, which turns out to be true when the data is analyzed in terms of a generalized mean field model [206]. This model incorporated a 6th order term in θ to account for the possibility of first order transition and a biquadratic coupling term ![]() to bring in transverse quadrupolar ordering (which is non-chiral in character). With terms relevant for the present case, the free energy is written as
to bring in transverse quadrupolar ordering (which is non-chiral in character). With terms relevant for the present case, the free energy is written as
(5.3) ![]()
yielding a simple relation between Ps and θ,
Here a, b, and c are the usual Landau coefficients with only the parameter a having a temperature dependence, χ is the susceptibility, C and Ω are the bilinear and biquadratic coupling coefficients between Ps and θ, and E is the applied electric field. In this model an all important parameter β ∝ C/Ω dictates the temperature dependence of the Ps/θ ratio.
Figure 5.25 Dependence of the spontaneous polarization (Ps) on reduced temperature in the absence of UV (top curve) and when the sample is illuminated with IUV = 4 mW/cm2 (bottom curve) for the antiferroelectric AFM mixture exhibiting PPE [ref. 205].
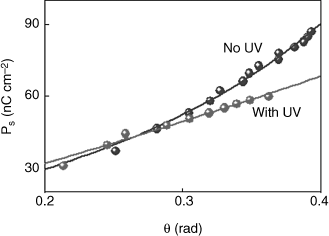
The polarization dependence on tilt angle with and without UV illumination is given in Figure 5.26, along with the fitting done using Eq. (5.4). To be noted is the feature that while the data collected without UV has a significant curvature, under UV illumination it is highly linear (In fact, a fit done to the expression for a straight line yields only a slightly poorer fit.). The C and Ω values obtained from the best fit of the data to Eq. (5.4) are 4.4 ± 0.1 × 107 V m−1 and 8.2 ± 0.4 × 1010 N m2 C−2 for the no UV data set and 5.2 ± 0.2 × 107 V m−1 and 1.8 ± 0.1 × 1010 N m2 C−2 for the data with UV illumination; these values are of the same order of magnitude as for other ferroelectric liquid crystals [205, 206]. The photoisomerization influences the non-chiral coefficient Ω (factor of 4 decrease) more than the chiral coefficient C (~17% increase), resulting in an increase of the ratio C/Ω by a factor of 5. In other words, UV illumination reduces the non-chiral aspect of the Ps–θ coupling, thus supporting the qualitative change in the behavior of the Ps vs. θ data from non-linear trend to a linear one as shown in Figure 5.26. These observations are further supported by the UV-intensity dependence of the thermal variation of the dielectric strength [207]. From a molecular level of view, it may be suggested that mutual steric interactions, and consequent mutual orientation directions of the steric dipoles of the guest photoactive molecules and the host non-photoactive molecules, are perhaps responsible for the experimental features.
Figure 5.26 Tilt angle dependence of polarization in the absence of and upon shining UV. Notice that the no UV data has a significant curvature, whereas the data obtained with UV is nearly linear. The fit to Eq. (5.4) (shown as solid lines) describes the data well in both cases [ref. 205].
5.3.7.2 A System with Bent-Core Molecules
Contrary to the earlier thinking, ferroelectricity has been observed in liquid crystals made of entirely achiral molecules, having a bent or banana shape [106–109]. The origin of spontaneous polarization in these systems is believed to be a combination of the following factors. These molecules have a highly polar character, are arranged in smectic layers in such a way that the dipoles point along a common direction within the layer and are tilted with respect to the layer normal. These factors can give rise to chiral layer symmetry, although the molecules themselves are achiral. Here again, the polarization Ps as well as the response time τ decrease upon photoisomerization of the guest EPH molecules in a host bent-core system; upon switching off the UV illumination, a rapid recovery to the equilibrium values is also observed (see Figure 5.27a) [208]. While ruling out the lowering of the transition temperature and the nanophase segregation mechanism as the cause for the reduction in Ps and τ (the response time), the results have been explained by considering the EPH molecules to remain in the smectic layer, even in the presence of the UV radiation. Since there is no additional volume cost, unlike for the host bent-core PHDBB molecules, the polar axis of the bent EPH molecules within one layer could point in either direction compared to the polar axis of the smectic layer. The antiferroelectric character of such an arrangement reduces the polar character of the layer, thereby reducing Ps values. In the tilt plane, i.e., the plane normal to twofold symmetry axis, the two Z molecules within one smectic layer can adopt a synclinic geometry. The disposition of the EPH molecules in this manner can perhaps be thought of as a “local racemization” (see Figure 5.27b). The behavior of the response time can also be explained with the above concept.
Figure 5.27 (a) Time dependence of the polarization upon UV illumination and subsequent switch OFF. The sample is kept at 4°C below the B2–Iso transition temperature. The time required for substantial change in the value seems to be comparable for both illumination and switch OFF processes. (b) Polar plane and tilt plane views of the bent-core host (red structures) and rod-like photoactive guest molecules (blue entities) in their Z form. In the right panel, crosses indicate polarization direction pointing towards, and the crosses, away from the reader. Reprinted with permission from ref. 208, Copyright 2001, American Institute of Physics.
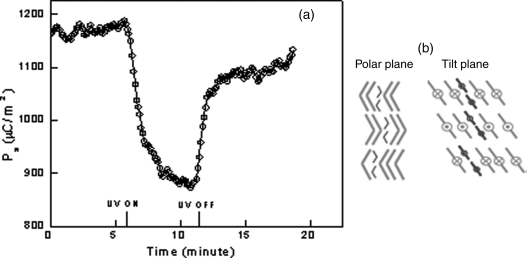
Photoinduced effect on the chiral azobenzene-based banana-shaped LCs (Figure 5.28) involving the N*-I transition and shift in selective reflection band position of the N* phase has been reported very recently [209]. A blue shift in the selective reflection band occurs when cis-isomers exist in low population; for a higher cis population isothermal transition to isotropic phase occurs. Using these materials as chiral dopants in the standard nematic mixture E7, their helical twisting power (HTP), given by β = 1/PC (where P is the helical pitch length, and C is the concentration of a chiral dopant) has been determined. The β values for trans isomers of 1 and 2 were found to be 4.5 μm−1 and 3.3 μm−1, respectively. A further lowering of β value was observed when cis-isomers are formed upon irradiation and thus, a light-induced hypsochromic shift results in the selective reflection band of the N* phase. These results are in agreement with the fact that a photoinduced shift in the N* pitch can also occur due to the difference in twisting ability of the trans and cis isomers of photochromic chiral additives [210], as also seen by others [140, 211]. Indeed, such photoinduced effects involving the shift in the selective reflection band of the N* phase have also been reported for a variety of photoactive compounds/dopants [21–27, 63–65, 78, 84–88, 140, 210, 211].
Figure 5.28 A schematic representation of chiral nematic (N*) phase with a photoresponsive, as well as thermally and electrically addressable, pitch P, composed of photosensitive optically active azobenzene-based banana-shaped compounds 1 and 2 [ref. 209].
5.4 Applications
5.4.1 Holography
Several photoactive polymeric LCs exhibiting N-I transitions have been examined in the context of dynamic holography phenomenon as it holds an immense promise in real-time image processing, phase conjugate mirrors, and other optical communication applications [31, 32, 49–52]. In particular, the important characteristics, viz., thermal stability of the stored information, resolution sensitivity, and storage capability have been evaluated; the high performance of these photoactive polymeric LC as a holographic recording medium has been revealed. Besides, a variety of low molar mass LCs [55–58] and LC/polymer composites [39, 40] have also been studied extensively in the context of both static and dynamic holographic gratings. However, it must be remarked here that Wendorff et al. pioneered the production of holograms in LC copolymers containing azobenzene moieties and mesogenic groups [33, 34, 61, 62].
Following the report of holographic device by Wendorff's group [33, 34], Ikeda and coworkers [35] demonstrated the potentials of N-I transition in optical image storage application by showing fast photochemical change (200 μs) both in low molar mass (BMAB and 8AB8) and polymeric (PA6AB2) nematic LCs (see Chart 5.5B). In this demonstration, the nematic phase of polymeric LCs, covered with a photomask, is made to undergo photoinduced N-I transition; the illuminated regions transform to the isotropic liquid phase, while the unexposed remain in the N phase. The sample then taken to the glass state retains the stored pattern, even though reverse isomerization (cis–trans) occurs, pointing to the fact that no phase transition takes place in the glass environment. This feature has been ascribed to the fact that while the segmental motions of polymer chains are possible, no macroscopic ordering (necessary for the isotropic liquid state to return to the equilibrium N phase) is realized. Following this different types of azobenzene–low molar mass LC composites [43, 44], azobenzene–polymeric LCs [45], polymeric LCs [49], and copolymers [50] have been prepared and their response were investigated.
The dual frequency feature of some nematogens has been exploited to create an optical storage device. The nematic phase of such materials exhibits a crossover frequency fc at which the dielectric anisotropy εa (=ε![]() − ε⊥) changes sign: from being positive for f < fc to negative for f > fc. Hence upon application of an electric field the molecules would feel a positive dielectric torque if f < fc and a negative torque if f > fc. In other words, above a certain magnitude of the electric field, the so-called Freedericksz threshold, the director prefers to be along or perpendicular to the electric field in the two cases, allowing the orientation (homeotropic/planar) to be controlled by the field itself, instead of appealing to surface forces. It has been demonstrated that such a configurational switching can be achieved at a constant frequency by illuminating the sample with actinic light [212]. The principle behind such a device is the substantial increase in the dielectric relaxation frequency fR (and thus indirectly in the crossover frequency fc), induced by the radiation (see Figure 5.29a). These changes are associated with strong changes in the transmitted intensity of the liquid crystal cell, which can be exploited to realize a photonic addressable device in which the bright and dark states are achieved without letting the material to transform to the isotropic phase, a highly desirable aspect particularly in view of enhancing the chemical stability of the liquid crystalline substance employed. Figure 5.29b is an optical storage device that is realized using this phenomenon. The sample was illuminated with UV light of 10 mW/cm2 intensity through a standard pattern (1951 USAF resolution pattern positive target) while maintaining a high frequency voltage across the cell.
− ε⊥) changes sign: from being positive for f < fc to negative for f > fc. Hence upon application of an electric field the molecules would feel a positive dielectric torque if f < fc and a negative torque if f > fc. In other words, above a certain magnitude of the electric field, the so-called Freedericksz threshold, the director prefers to be along or perpendicular to the electric field in the two cases, allowing the orientation (homeotropic/planar) to be controlled by the field itself, instead of appealing to surface forces. It has been demonstrated that such a configurational switching can be achieved at a constant frequency by illuminating the sample with actinic light [212]. The principle behind such a device is the substantial increase in the dielectric relaxation frequency fR (and thus indirectly in the crossover frequency fc), induced by the radiation (see Figure 5.29a). These changes are associated with strong changes in the transmitted intensity of the liquid crystal cell, which can be exploited to realize a photonic addressable device in which the bright and dark states are achieved without letting the material to transform to the isotropic phase, a highly desirable aspect particularly in view of enhancing the chemical stability of the liquid crystalline substance employed. Figure 5.29b is an optical storage device that is realized using this phenomenon. The sample was illuminated with UV light of 10 mW/cm2 intensity through a standard pattern (1951 USAF resolution pattern positive target) while maintaining a high frequency voltage across the cell.
Figure 5.29 (a) Dielectric dispersion scans taken in the nematic phase exhibiting the dual frequency character before (open triangles), and with steady-state UV radiation (open circles). Notice that the relaxation frequency (indicated by the arrows) is higher for the UV-on case. After the radiation is switched off, the original value is recovered, as shown by the data indicated with filled squares. For the sake of clarity, the solid line representing the fit to the Havriliak–Negami equation is shown only for the UV-on data. (b) Optical storage device realized using the dual frequency principle. The sample was kept at room temperature and illuminated with UV radiation through a photomask (a standard 2″ 1951 USAF resolution pattern positive target). The dark regions are where the molecules are in the homeotropic alignment caused by the passing of the UV and the bright regions are where the radiation is masked. Reprinted with permission from ref. 212, Copyright 2005, American Institute of Physics.

5.4.2 Soft Actuators
In recent times, the photoinduced N to I transformation has been also used in developing soft actuators derived from liquid crystalline elastomers (LCEs) [27, 90–95, 213, 214]. The possibility of coupling between orientational order and mechanical properties in LCEs, pointed out by de Gennes [91, 92], has potential application as artificial muscles and mechanically tunable optical elements [215]. In fact, experimentally, a spontaneous contraction along the director axis of the nematic LCE films upon heating towards the N-I transition has been observed [93]. Notably, the deformation in the shape up to 300% has been achieved in nematic LCE films derived from side-chain siloxane polymers [94]. Developing on the concept of photomechanical effects of a monolayer consisting of polyamides [216], large and reversible deformation of LCEs driven by actinic light have been demonstrated; the gradient of the cross-linking density, polarization direction of actinic light, the initial alignment of the LC molecules, etc. are found to control the associated motions of such LCE films [95, 100, 217]. Detailed mechanical characterization of such films has also been performed recently [218].
The occurrence of this photomechanical effect has been attributed to the decrease in the order parameter caused by trans–cis isomerization. For example, Ikeda's group [95–98] has accomplished photoinduced three-dimensional deformation of the azobenzene-based nematic LCEs swollen in suitable solvents or heated above their Tg in air. They have been successful in developing a single film capable of repeatedly bending precisely along the directed direction [27, 90, 99]. An example of such a photochemical effect is shown in Figure 5.30. Likewise, a number of investigations have been reported to show that the LCEs derived from azobenzene moiety, in response to the illuminated light, exhibit remarkable changes in shape and volume where the light energy directly transforms into the mechanical energy. Photoswitchable ferroelectric LCEs have also been realized paving the way to control the shape changes through optical (isomerization) as well as electric (Piezo effect) means in addition to the more established thermal path [219].
Figure 5.30 (a) Demonstration of photocontraction of a cross-linked polymer liquid crystal containing azobenzene, in which the bending direction of the film is manipulated by the orientation of linearly polarized light in the UV region (inducing contraction) and visible light (recovery of the original shape). Reprinted with permission from ref. 100, Copyright 2003, Nature publishing group. (b) Schematic to illustrate the proposed mechanism governing the photocontraction. Reprinted with permission from ref. 90, Copyright 2006, John Wiley & Sons. (See the color version of this figure in Color Plates section.)
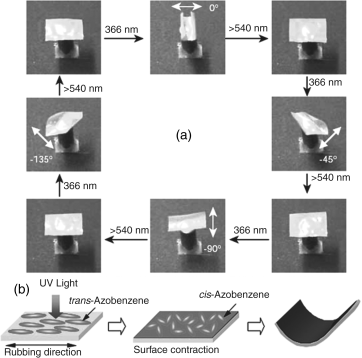
5.5 Summary and Outlook
Over the years, analogous to the parameters such as temperature, concentration of the solvent, and pressure, the significance of light of specific wavelength (actinic light) in driving liquid crystal phase transitions has been well demonstrated. Caused by the ability of light to alter/stabilize a particular thermodynamic phase via the photoisomerization, and consequent shape change of the constituent molecules, such a phenomenon provides an interesting tool to investigate condensed matter from a new dimension. A beautiful illustration of nature utilizing such photoinduced effects is that of the retinal protein rhodopsin that enables vision. In the artificial world this effect is perhaps best demonstrated by studies involving photosensitive organic molecules. This has resulted in a number of photoactive materials, for example, azobenzene or its derivatives, being employed either in pure form or as a guest component in guest–host mixtures, to investigate the stimulus provided by the photoresponsive part on the response of the entire system. In liquid crystalline systems, a diverse variety of photoinduced phase transitions have been observed involving achiral as well as chiral materials, nematic, smectic, columnar, and blue phases. A detailed account of these photo-stimulated, but isothermal transformations provided in this chapter illustrates their significance in both basic research and applied sciences. Given the fact that phase transition among the mesophases and their transition to isotropic phase bring about the changes in their optical properties effectively, the usage of light-driven phase transitions for such purposes continues to be a fascinating field of exploration. Notably, in recent years, the effects of parameters such as surface fields, electric field, elevated pressure, anisotropy reducing component, and magnitude of orientational order on the photoinduced nematic–isotropic transition have been well studied to signify their importance in technological aspects. Likewise, dynamic self-assembly of a layered phase induced and stabilized by actinic light, tilt susceptibility of chiral smectic system, primary and secondary photo ferroelectric effects in an antiferroelectric LC, etc. focusing on the static and dynamic properties highlight the fact that the actinic light can be used as a new tool to study phase transitions and the associated critical phenomena that could also bring about effects that are not seen in equilibrium situations. Most importantly, two applications originating from the photoisomerization, viz., optical storage and photomechanical effect have been well verified. In addition, several non-display applications can be expected to emerge, provided new photochromic molecules and photo-induced phase transitions with promising static and dynamic features evolve.
1. S. Chandrasekhar, Liquid Crystals, 2nd Edition, Cambridge University Press, 1994.
2. P. G. de Gennes and J. Prost. The Physics of Liquid Crystals, Oxford University Press, Oxford, 1994.
3. D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess, and V. Vill. Handbook of Liquid Crystals, Vol. 1: Fundamentals, Wiley-VCH, Germany, 1998.
4. D. Demus. One hundred years of liquid-crystal chemistry: thermotropic liquid crystals with conventional and unconventional molecular structure. Liq. Cryst. 1989, 5, 75–110.
5. C. Tschierske. Liquid crystalline materials with complex mesophase morphology. Curr. Opin. Colloid Interface Sci. 2002, 7, 69–80.
6. C. Tschierske. Micro-segregation, molecular shape and molecular topology – partners for the design of liquid crystalline materials with complex mesophase morphologies. J. Mater. Chem. 2001, 11, 2647–2671.
7. J. W. Goodby, I. M. Saez, S. J. Cowling, V. Gortz, M. Draper, A. W. Hall, S. Sia, G. Cosquer, S.-E. Lee, and E. P. Raynes. Transmission and amplification of information and properties in nanostructured liquid crystals. Angew. Chem. Int. Ed. 2008, 47, 2754–2787.
8. IUPAC. Compendium of Chemical Terminology, 2nd Edition (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford, 1997. XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. doi: 10.1351/goldbook.
9. H. Rau. Photoisomerization of azobenzenes. In: F. J. Rabeck,Ed., Photochemistry and Photophysics, vol. 2, CRC Press, Boca Raton, FL, 1990, Chapter 4.
10. N. Tamai and H. Miyasaka. Ultrafast dynamics of photochromic systems. Chem. Rev. 2000, 100, 1875–1890.
11. T. Ishikawa, T. Noro, and T. Shoda. Theoretical study on the photoisomerization of azobenzene. J. Chem. Phys. 2001, 115, 7503–7512.
12. K. G. Yager and C. J. Barrett. Novel photo-switching using azobenzene functional materials. J. Photochem. Photobiol. A: Chem. 2006, 182, 250–261.
13. H. Rau, In: H. Dürr and H. Bouas-Laurent,Eds., Photochromism – Molecules and Systems, vol. 1, Elsevier, Amsterdam, 1990, Chapter 4, and references cited therein.
14. H. Stegemeyer. On the mechanism of photochemical cis–trans isomerization. J. Phys. Chem. 1962, 66, 2555–2560.
15. M. Dumont, S. Hosotte, G. Froc, and Z. Sekkat. Orientational manipulation of chromophores through photoisomerization. Proc. SPIE 1994 2042, 2–13.
16. I. Lednev, T. Q. Ye, R. Hester, and J. Moore. Femtosecond time-resolved UV-visible absorption spectroscopy of trans-azobenzene in solution. J. Phys. Chem. 1996, 100, 13338–13341.
17. T. V. Galstyan, B. Saad, and M. M. Denariez-Roberge. Excitation transfer from azo dye to nematic host during photoisomerization. J. Chem. Phys. 1997, 107, 9319–9325.
18. N. Biswas and S. Umapathy. Resonance Raman study of the solvent dynamics for ultrafast charge transfer transition in 4-nitro-4′-dimethylamino-azobenzene. J. Chem. Phys. 2003, 118, 5526–5536.
19. C. R. Crecca and A. E. Roitberg. Theoretical study of the isomerization mechanism of azobenzene and disubstituted azobenzene derivatives. J. Phys. Chem. A 2006, 110, 8188–8203.
20. Y. Douab, Y. Hua, S. Yuanac, W. Wua, and H. Tang. Detailed mechanism of trans–cis photoisomerization of azobenzene studied by semiclassical dynamics simulation. Mol. Phys. 2009, 107, 181–190.
21. T. Ikeda. Photomodulation of liquid crystal orientations for photonic applications. J. Mater. Chem. 2003, 13, 2037–2057.
22. S. Das and R. Davis. Photoresponsive liquid crystals. Proc. Indian Natl. Sci. Acad. 2003, 69A, 109–122.
23. Y. Yu and T. Ikeda. Alignment modulation of azobenzene-containing liquid crystal systems by photochemical reactions. J. Photochem. Photobiol. C: Photochem. Rev. 2004, 5, 247–265.
24. S. K. Prasad, G. G. Nair, K. L. Sandhya, and D. S. Shankar Rao. Photoinduced phase transitions in liquid crystalline systems. Curr. Sci. 2004, 86, 815–823.
25. S. K. Prasad, G. G. Nair, G. Hegade, K. L. Sandhya, D. S. Shankar Rao, C. V. Lobo, and C. V. Yelamaggad. Photoinduced effects in nematic liquid crystals. Phase Transitions 2005, 78, 443–455.
26. S. K. Prasad, G. G. Nair, and D. S. S. Rao. Photoinduced phase transitions. Liq. Cryst. 2009, 36, 705–716.
27. H. Yu and T. Ikeda. Photocontrollable liquid-crystalline actuators. Adv. Mater. 2011, 23, 2149–2180.
28. E. Sackmann. Photochemically induced reversible color changes in cholesteric liquid crystals. J. Am. Chem. Soc. 1971, 93, 7088–7090.
29. W. E. Haas, K. F. Nelson, J. E. Adams, and G. A. Dir. UV-imaging with nematic chlorostilbenes. J. Electrochem. Soc. 1974, 121, 1667–1669.
30. K. F. Nelson, W. E. Haas, and J. E. Adams. Identification of the UV irradiation product in nematic chlorostilbene imaging processes. J. Electrochem. Soc. 1975, 122, 1564–1565.
31. G. Pelzl. Transformation of nematic into isotropic phase by photochemical isomerization. Z. Chem. 1977, 17, 294–295.
32. S. Tazuke, S. Kurihara, and T. Ikeda. Amplified image recording in liquid crystal media by means of photochemically triggered phase transition. Chem. Lett. 1987, 911–914.
33. M. Eich, J. H. Wendorff, B. Reck, and H. Ringsdorf. Reversible digital and holographic optical storage in polymeric liquid crystals. Macromol. Chem. Rapid Commun. 1987, 8, 59–63.
34. M. Eich and J. H. Wendorff. Erasable holograms in polymeric liquid crystals. Macromol. Chem. Rapid Commun. 1987, 8, 467–471.
35. T. Ikeda and O. Tsutsumi. Optical switching and image storage by means of azobenzene liquid-crystal films. Science, 1995, 268, 1873–1875.
36. O. Tsutsumi, T. Shiono, T. Ikeda, and G. Galli. Photochemical phase transition behavior of nematic liquid crystals with azobenzene moieties as both mesogens and photosensitive chromophores. J. Phys. Chem. B 1997, 101, 1332–1337.
37. A. Shishido, O. Tsutsumi, A. Kanazawa, T. Shiono, and T. Ikeda. Distinct photochemical phase transition behavior of azobenzene liquid crystals evaluated by reflection-mode analysis. J. Phys. Chem. B 1997, 101, 2806–2810.
38. A. Shishido, O. Tsutsumi, A. Kanazawa, T. Shiono, T. Ikeda, and N. Tamai. Rapid optical switching by means of photoinduced change in refractive index of azobenzene liquid crystals detected by reflection-mode analysis. J. Am. Chem. Soc. 1997, 119, 7791–7796.
39. S. Kurihara, T. Ikeda, and S. Tazuke. Photochemically induced isothermal phase transition in liquid crystals. Effect of interaction of photoresponsive molecules with matrix mesogens. Mol. Cryst. Liq. Cryst. 1990, 178, 117–132.
40. C. Ruslim and K. Ichimura. Z-Isomers of azobenzenes highly compatible with liquid crystals. Chem. Lett. 1998, 789–790.
41. C. H. Legge and G. R. Mitchell. Photo-induced phase transitions in azobenzene-doped liquid crystals. J. Phys. D: Appl. Phys. 1992, 25, 492–499.
42. J.-H. Sung, S. Hirano, O. Tsutsumi, A. Kanazawa, T. Shiono, and T. Ikeda. Dynamics of photochemical phase transition of guest/host liquid crystals with an azobenzene derivative as a photoresponsive chromophore. Chem. Mater. 2002, 14, 385–391.
43. S. Kurihara, T. Ikeda, T. Sasaki, H.-B. Kim, and S. Tazuke. Time-resolved observation of isothermal phase transition of liquid crystals triggered by photochemical reaction of dopant. J. Chem. Soc., Chem. Commun. 1990, 1751–1752.
44. S. Kurihara, T. Ikeda, T. Sasaki, H.-B. Kim and S. Tazuke. Time-resolved observation of isothermal transition of liquid crystals induced by photoisomerization of azobenzene dopant. Mol. Cryst. Liq. Cryst. 1991, 195, 251–263.
45. T. Ikeda, T. Sasaki, and H.-B. Kim. Intrinsic response of polymer liquid crystals in photochemical phase transition. J. Phys. Chem. 1991, 95, 509–511.
46. H.-K. Lee, A. Kanazawa, T. Shiono, and T. Ikeda. All-optically controllable polymer/liquid crystal composite films containing the azobenzene liquid crystal. Chem. Mater. 1998, 10, 1402–1407.
47. S. Xie, A. Natansohn, and P. Rochoni. Recent developments in aromatic azo polymers research. Chem. Mater. 1993, 5, 403–411.
48. O. Tsutsumi, Y. Demachi, A. Kanazawa, T. Shiono, T. Ikeda, and Y. Nagase. Photochemical phase-transition behavior of polymer liquid crystals induced by photochemical reaction of azobenzenes with strong donor–acceptor pairs. J. Phys. Chem. B 1998, 102, 2869–2874.
49. O. Tsutsumi, T. Kitsunai, A. Kanazawa, T. Shiono, and T. Ikeda. Photochemical phase transition behavior of polymer azobenzene liquid crystals with electron-donating and accepting substituents at the 4, 4′-positions. Macromolecules 1998, 31, 355–359.
50. T. Sasaki, T. Ikeda, and K. Ichimura. Time-resolved observation of photochemical phase transition in polymer liquid crystals. Macromolecules 1992, 25, 3807–3811.
51. M. Hasegawa, T. Yamamoto, A. Kanazawa, T. Shiono, and T. Ikeda. A dynamic grating using a photochemical phase transition of polymer liquid crystals containing azobenzene derivatives. Adv. Mater. 1999, 11, 675–677.
52. T. Yamamoto, M. Hasegawa, A. Kanazawa, T. Shiono, and T. Ikeda. Phase-type gratings formed by photochemical phase transition of polymer azobenzene liquid crystals: enhancement of diffraction efficiency by spatial modulation of molecular alignment. J. Phys. Chem. B 1999, 103, 9873–9878.
53. M. Hasegawa, T. Yamamoto, A. Kanazawa, T. Shiono, and T. Ikeda. Photochemically induced dynamic grating by means of side chain polymer liquid crystals. Chem. Mater. 1999, 11, 2764–2769.
54. T. Yamamoto, M. Hasegawa, A. Kanazawa, T. Shiono, and T. Ikeda. Holographic gratings and holographic image storage via photochemical phase transitions of polymer azobenzene liquid crystal films. J. Mater. Chem. 2000, 10, 337–342.
55. R. Ortler, C. Brauchle, A. Miller, and G. Riepl. Reversible holographic-optical data storage in cholesteric liquid-crystalline siloxanes. Macromol. Chem. Rapid Commun. 1989, 10, 189–194.
56. G. P. Wiederrecht, B. A. Yoon, and M. R. Wasielewski. High photorefractive gain in nematic liquid crystals doped with electron donor and acceptor molecules. Science 1995, 270, 1794–1797.
57. G. P. Wiederrecht, B. A. Yoon, W. A. Svec, and M. R. Wasielewski. Photorefractivity in nematic liquid crystals containing electron donor−acceptor molecules that undergo intramolecular charge separation. J. Am. Chem. Soc. 1997, 119, 3358–3364.
58. T. Sasaki, A. Katsuragi, and K. Ohno. Spontaneous polarization vector reorientation photorefractive effect in dye-doped ferroelectric liquid crystals. J. Phys. Chem. B 2002, 106, 2520–2525.
59. F. Simoni, F. Bloisi, and L. Vicari. Transient amplitude grating in polymer dispersed liquid crystals. Mol. Cryst. Liq. Cryst. 1992, 223, 169–179.
60. T. Ubukata, T. Seki, and K. Ichimura. Surface relief gratings in host–guest supramolecular materials. Adv. Mater. 2000, 12, 1675–1678.
61. J. H. Wendorff and M. Eich. Nonlinear optical phenomenon in liquid crystalline side chain polymers. Mol. Cryst. Liq. Cryst. 1989, 169, 133–166.
62. K. Anderle and J. H. Wendorff. Holographic recording, using liquid crystalline side chain polymers. Mol. Cryst. Liq. Cryst. 1994, 243, 51–75.
63. B. L. Feringa, W. F. Jager, and B. de Lange. Organic materials for reversible optical data storage. Tetrahedron 1993, 49, 8267–8310.
64. B. L. Feringa, R. A. van Delden, N. Koumura, and E. M. Geertsema. Chiroptical molecular switches. Chem. Rev. 2000, 100, 1789–1816.
65. R. Eelkema and B. L. Feringa. Amplification of chirality in liquid crystals. Org. Biomol. Chem. 2006, 4, 3729–3745.
66. A. Chanishvili, G. Chilaya, G. Petriashvili, and D. Sikharulidze. Light induced effects in cholesteric mixtures with a photosensitive nematic host. Mol. Cryst. Liq. Cryst. 2004, 409, 209–218.
67. A. Chanishvili, G. Chilaya, G. Petriashvili, and P. J. Collings. Trans–cis isomerization and the blue phases. Phys. Rev. E 2005, 71, 051705-1–051705-5.
68. D. Aronzon, E. P. Levy, P. J. Collings, A. Chanishvill, G. Chilaya, and G. Petriashvili. Trans–cis isomerization of an azoxybenzene liquid crystal. Liq. Cryst. 2007, 34, 707–718.
69. S. Kurihara, T. Ikeda, S. Tazuke, and J.-e. Seto. Isothermal phase transition of liquid crystals induced by photoisomerization of doped spiropyrans. J. Chem. Soc., Faraday Trans. 1991, 87, 3251–3254.
70. H. Allinson and H. F. Gleeson. Dielectric permittivity properties of a fulgide dye guest–host liquid crystal. Liq. Cryst. 1995, 19, 421–425.
71. H. Allinson and H. F. Gleeson. Variations in the elastic constants of a fulgide-doped liquid crystal system. J. Mater. Chem. 1995, 5, 2139–2144.
72. M. Irie, K. Sakemura, M. Okinaka, and K. Uchida. Photochromism of dithienylethenes with electron-donating substituents. J. Org. Chem. 1995, 60, 8305.
73. M. Irie and K. Uchida. Synthesis and properties of photochromic diarylethenes with heterocyclic aryl groups. Bull. Chem. Soc. Jpn. 1998, 71, 985–996.
74. T. Yamaguchi, T. Inagawa, H. Nakazumi, S. Irie, and M. Irie. Photoswitching of helical twisting power of a chiral diarylethene dopant: pitch change in a chiral nematic liquid crystal. Chem. Mater. 2000, 12, 869–871.
75. C. Denekamp and B. L. Feringa. Optically active diarylethenes for multimode photoswitching between liquid-crystalline phases. Adv. Mater. 1998, 10, 1080–1082.
76. K. Matsuda and M. Irie. Diarylethene as a photoswitching unit. J. Photochem. Photobiol. C: Photochem. Rev. 2004, 5, 169–182.
77. H. Tian and S. Yang. Recent progresses on diarylethene based photochromic switches. Chem. Soc. Rev. 2004, 33, 85–97.
78. K. Rameshbabu, A. Urbas, and Q. Li. Synthesis and characterization of thermally irreversible photochromic cholesteric liquid crystals. J. Phys. Chem. B 2011, 115, 3409–3415.
79. V. A. Mallia, M. George, and S. Das. Photochemical phase transition in hydrogen-bonded liquid crystals. Chem. Mater. 1999, 11, 207–208.
80. R. Brettle, D. A. Dunmur, N. J. Hindley, and C. M. Marson. Synthesis of new liquid crystalline compounds based on 1,4-diarylbuta-1,3-dienes. J. Chem. Soc., Perkin Trans. 1 1993, 775–781.
81. S. Das, S. Varghese, and N. S. S. Kumar. Butadiene-based photoresponsive soft materials. Langmuir 2010, 26, 1598–1609.
82. M. N. Pivnenko, V. V. Vashchenko, A. I. Krivoshey, L. A. Kutulya, and J. W. Goodby. New chiral ether derivatives of 2-arylidene-p-menthane-3-ones as components of induced ferroelectric systems. Mol. Cryst. Liq. Cryst. 2001, 364, 557–565.
83. T. G. Drushlyak, A. Kutulya, N. S. Pivnenko, N. I. Shkolnikov, and V. V. Vashchenko. New chiral esters, diastereomeric 2-(4-carboxybenzylidene)-p-menthane-3-one derivatives, as components of LC systems with induced helical structure. Mol. Cryst. Liq. Cryst. 2001, 364, 691–701.
84. Q. Li, L. Green, N. Venkataraman, I. Shiyanovskaya, A. Khan, A. Urbas, and J. W. Doane. Reversible photoswitchable axially chiral dopants with high helical twisting power. J. Am. Chem. Soc. 2007, 129, 12908–12909.
85. T. J. White, R. L. Bricker, L. V. Natarajan, N. V. Tabiryan, L. Green, Q. Li, and T. J. Bunning. Phototunable azobenzene cholesteric liquid crystals with 2000 nm range. Adv. Funct. Mater. 2009, 19, 3484–3488.
86. L. Green, Y. Li, T. White, A. Urbas, T. Bunning, and Q. Li. Light-driven molecular switches with tetrahedral and axial chirality. Org. Biomol. Chem. 2009, 7, 3930–3933.
87. T. J. White, R. L. Bricker, L. V. Natarajan, V. P. Tondiglia, L. Green, Q. Li, and T. J. Bunning. Electrically switchable, photoaddressable cholesteric liquid crystal reflectors. Opt. Express 2010, 18, 173–178.
88. J. Ma, Y. Li, T. White, A. Urbas, and Q. Li. Light-driven nanoscale chiral molecular switch: reversible dynamic full range colour phototuning. Chem. Commun. 2010, 46, 3463–3465.
89. T. V. Truong, C.-Y. Chen, N. V. Tabiryan, and Y. Ron Shen. Phase transition induced by two-photon absorption in azobenzene liquid crystals. J. Opt. Soc. Am. B 2007, 24, 2623–2626.
90. Y. Yu and T. Ikeda. Soft actuators based on liquid-crystalline elastomers. Angew. Chem. Int. Ed. 2006, 45, 5416–5418.
91. P.G. de Gennes. Possibilities offered by polymer crosslinking in the presence of a liquid crystal. Phys. Lett. A 1969, 28, 725–726.
92. P. G. de Gennes, M. Hebert, and R. Kant. Artificial muscles based on nematic gels. Macromol. Symp. 1997, 113, 39–49.
93. I. Kundler and H. Finkelmann. Director reorientation via stripe-domains in nematic elastomers: influence of cross-link density, anisotropy of the network and smectic clusters. Macromol. Chem. Phys. 1998, 199, 677–686.
94. S. M. Clarke, A. Hotta, A. R. Taj-bakhsh, and E. M. Terentjev. Effect of cross-linker geometry on equilibrium thermal and mechanical properties of nematic elastomers, Phys. Rev. E 2001, 64, 061702-1–061702-8.
95. H. Finkelmann, E. Nishikawa, G. G. Pereira, and M. Warner. A new opto-mechanical effect in solids. Phys. Rev. Lett. 2001, 87, 015501.
96. P. M. Hogan, A. R. Tajbakhsh, and E. M. Terentjev. UV manipulation of order and macroscopic shape in nematic elastomers. Phys. Rev. E 2002, 65, 041720.
97. M.-H. Li, P. Keller, B. Li, X. Wang and M. Brunet. Light-driven side-on nematic elastomer actuators. Adv. Mater. 2003, 15, 569–572.
98. T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi, and A. Kanazawa. Anisotropic bending and unbending behavior of azobenzene liquid-crystalline gels by light exposure. Adv. Mater. 2003, 15, 201–205.
99. Y. Yu, M. Nakano, A. Shishido, T. Shiono, and T. Ikeda. Effect of cross-linking density on photoinduced bending behavior of oriented liquid-crystalline network films containing azobenzene. Chem. Mater. 2004, 16, 1637–1643.
100. Y. Yu, M. Nakano, and T. Ikeda. Directed bending of a polymer film by light. Nature 2003, 425, 145.
101. M. Kondo, Y. Yu, and T. Ikeda. How does the initial alignment of mesogens affect the photoinduced bending behavior of liquid-crystalline elastomers? Angew. Chem. Int. Ed. 2006, 45, 1378–1382.
102. M. Camacho-Lopez, H. Finkelmann, P. Palffy-Muhoray, and M. Shelley. Fast liquid-crystal elastomer swims into the dark. Nat. Mater. 2004, 3, 307–310.
103. K. D. Harris, R. Cuypers, P. Scheibe, C. L. van Oosten, C. W. M. Bastiaansen, J. Lub, and D. J. Broer. Large amplitude light-induced motion in high elastic modulus polymer actuators. J. Mater. Chem. 2005, 15, 5043–5048.
104. M. M. Green, R. J. M. Nolte, and E. W. Meijer,Eds., Topics in Stereochemistry, Vol. 24: Materials-Chirality, Wiley-VCH, New York, 2003.
105. J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte, and N. A. J. M. Sommerdijk. Chiral architectures from macromolecular building blocks. Chem. Rev. 2001, 101, 4039–4070.
106. R. A. Reddy and C. Tschierske. Bent-core liquid crystals: polar order, superstructural chirality and spontaneous desymmetrisation in soft matter systems. J. Mater. Chem. 2006, 16, 907–961.
107. H. Takezoe and Y. Takanishi. Bent-core liquid crystals: their mysterious and attractive world. Jpn. J. Appl. Phys. 2006, 45, 597–625.
108. A. Jakli, C. Bailey, and J. Harden, Physical properties of banana liquid crystals. In: A. Ramamoorthy,Ed., Thermotropic Liquid Crystals. Recent Advances, Springer, Netherlands, 2007, Chapter 2.
109. D. R. Link, G. Natale, R. Shao, J. E. Maclennan, N. A. Clark, E. Korblova, and D. M. Walba. Spontaneous formation of macroscopic chiral domains in a fluid smectic phase of achiral molecules. Science 1997, 278, 1924–1927.
110. M. Avalos, R. Babiano, P. Cintas, J. L. Jimenez, J. C. Palacios and L. D. Barron. Absolute asymmetric synthesis under physical fields: facts and fictions. Chem. Rev. 1998, 98, 2391–2404.
111. R. Maria Tejedor, L. Oriol, J. L. Serrano, and T. Sierra. Chiral photochemical induction in liquid crystals. J. Mater. Chem. 2008, 18, 2899–2908.
112. M. Suarez and G. B. Schuster. Photoresolution of an axially chiral bicyclo [3.3.0]octan-3-one: phototriggers for a liquid-crystal-based optical switch. J. Am. Chem. Soc. 1995, 117, 6732–6738.
113. M. Zhang and G. B. Schuster. Photoracemization of optically active 1,l′-binaphthyl derivatives: light-initiated conversion of cholesteric to compensated nematic liquid crystals. J. Phys. Chem. 1992, 96, 3063–3067.
114. K. S. Burnham and G. B. Schuster. A search for chiral photochromic optical triggers for liquid crystals: photoracemization of 1,1′-binaphthylpyran through a transient biaryl quinone methide intermediate. J. Am. Chem. Soc. 1998, 120, 12619–12625.
115. B. L. Feringa, N. P. M. Huck, and H. A. van Dorenl. Chiroptical switching between liquid crystalline phases. J. Am. Chem. Soc. 1995, 117, 9929–9930.
116. N. P. M. Huck, W. F. Jager, B. de Lange, and B. L. Feringa. Dynamic control and amplification of molecular chirality by circular polarized light. Science 1996, 273, 1686–1688.
117. K. S. Burnham and G. B. Schuster. Transfer of chirality from circularly polarized light to a bulk material property: propagation of photoresolution by a liquid crystal transition. J. Am. Chem. Soc. 1999, 121, 10245–10246.
118. K. L. Stevenson, J. F. Verdieck, Partial photoresolution. Preliminary studies on some oxalato complexes of chromium (III). J. Am. Chem. Soc. 1968, 90, 2974–2975.
119. K. L. Stevenson. Partial photoresolution. III. Tris(acetylacetonato) chromium(III) system. J. Am. Chem. Soc. 1972, 94, 6652–6654.
120. B. Norden. Optical activity developed by preferential racemization of one enantiomer in racemic Cr(III) (ox)33− induced by irradiation with circularly polarized light. Acta Chem. Scand. 1970, 24, 349–351.
121. B. Norden. Optical activity in racemic chromium(III) tartrate solution induced by circularly polarized irradiation. Chem. Lett. 1977, 13, 355–362.
122. J. G. Radziszewski, J. W. Downing, M. Jawdosiuk, P. Kovacic, and J. Michl. 4-Azahomoadamant-3-ene: spectroscopic characterization and photoresolution of a highly reactive strained bridgehead imine. J. Am. Chem. Soc. 1985, 107, 594–603.
123. N. Tamaoki and M. Wada. Dynamic control of racemization rate through E–Z photoisomerization of azobenzene and subsequent partial photoresolution under circular polarized light. J. Am. Chem. Soc. 2006, 128, 6284–6285.
124. M. Mathews and N. Tamaoki. Reversibly tunable helicity induction and inversion in liquid crystal self-assembly by a planar chiroptic trigger molecule. Chem. Commun. 2009, 3609–3611.
125. S.-W. Choi, T. Izumi, Y. Hoshino, Y. Takanishi, K. Ishikawa, J. Watanabe, and H. Takezoe. Circular-polarization-induced enantiomeric excess in liquid crystals of an achiral, bent-shaped mesogen. Angew. Chem. Int. Ed. 2006, 45, 1382–1385.
126. F. Vera, R. M. Tejedor, P. Romero, J. Barberá, M. B. Ros, J. L. Serrano, and T. Sierra. Light-driven supramolecular chirality in propeller-like hydrogen-bonded complexes that show columnar mesomorphism. Angew. Chem. Int. Ed. 2007, 46, 1873–1877.
127. L. Nikolova, T. Todorov, M. Ivanov, F. Andruzzi, S. Hvilsted, and P. S. Ramanujam. Photoinduced circular anisotropy in side chain azobenzene polyesters. Opt. Mater. 1997, 8, 255–258.
128. L. Nedelchev, A. Matharu, L. Nikolova, S. Hvilsted, and P. S. Ramanujam. Propagation of polarized light through azobenzene polymer films. Mol. Cryst. Liq. Cryst. 2002, 375, 563–575.
129. R. M. Tejedor, L. Oriol, J. L. Serrano, F. Partal Urena, and J. J. L. Gonzalez. Photoinduced chiral nematic organization in an achiral glassy nematic azopolymer. Adv. Funct. Mater. 2007, 17, 3486–3492.
130. J. Garcia-Amoros, A. Szymczyka, and D. Velasco. Nematic-to-isotropic photo-induced phase transition in azobenzene-doped low-molar liquid crystals. Phys. Chem. Chem. Phys. 2009, 11, 4244–4250.
131. G. Joly, A. Anakkar, M. Ismaili, P. Cluzeau, N. Isaert, and H. T. Nguyen. Chiral azobenzene liquid crystals under illumination: thickness influence and spontaneous polarization variations. Ferroelectrics 2002, 277, 67–74.
132. G. Joly, A. Anakkar, and H. T. Nguyen. Light induced shifts of ferroelectric mesophase transitions. Liq. Cryst. 1999, 26, 1251–1255.
133. Y. Boussoualem, M. Ismaili, J. M. Buisine, C. Binet, G. Joly, and H. T. Nguyen. Dielectric and electro-optical properties of a photosensitive liquid crystal. Liq. Cryst. 2009, 36, 899–905.
134. R. Davis, V. A. Mallia, and S. Das. Reversible photochemical phase transition behavior of alkoxy-cyano-substituted diphenylbutadiene liquid crystals. Chem. Mater. 2003, 15, 1057–1063.
135. R. K. Vijayaraghavan, S. Abraham, D. S. Shankara Rao, S. K. Prasad, and S. Das. Light induced generation of stable blue phase in photoresponsive diphenylbutadiene based mesogen. Chem. Commun. 2010, 46, 2796–2798.
136. H.-Y. Liu, C.-T. Wang, C.-Y. Hsu, and T.-H. Lin. Optically tuneable blue phase photonic band gaps. Appl. Phys. Lett. 2010, 96, 121103-1–121103-3.
137. H.-Y. Liu, C.-T. Wang, C.-Y. Hsu, and T.-H. Lin. Photonic bandgaps controllable blue phase liquid crystal. Proc. SPIE 2011, 7955, 795506-1–795506-6.
138. T. Yamamoto, I. Nishiyama, and H. Yokoyama. Photo-control of chirality-induced liquid-crystalline 3D structures. Ferroelectrics 2008, 365, 39–47.
139. C. V. Yelamaggad, G. Shanker, U. S. Hiremath, and S. K. Prasad. Cholesterol-based nonsymmetric liquid crystal dimers: an overview. J. Mater. Chem. 2008, 18, 2927–2949.
140. N. Tamaoki, Y. Aoki, M. Moriyama, and M. Kidowaki. Photochemical phase transition and molecular realignment of glass-forming liquid crystals containing cholesterol/azobenzene dimesogenic compounds. Chem. Mater. 2003, 15, 719–726.
141. V. A. Mallia and N. Tamaoki. Photoresponsive vitrifiable chiral dimesogens: photo-thermal modulation of microscopic disordering in helical superstructure and glass-forming properties. J. Mater. Chem. 2003, 13, 219–224.
142. V. A. Mallia and N. Tamaoki. Photochemically driven smectic–cholesteric phase transition in an inherently photoactive dimesogen. Chem. Mater. 2003, 15, 3237–3239.
143. V. A. Mallia and N. Tamaoki. Photoactive dimesogen having different pathways of light driven phase transitions at different temperatures. Chem. Commun. 2004, 2538–2539.
144. S. Abraham, V. A. Mallia, K. V. Ratheesh, N. Tamaoki, and S. Das. Reversible thermal and photochemical switching of liquid crystalline phases and luminescence in diphenylbutadiene-based mesogenic dimers. J. Am. Chem. Soc. 2006, 128, 7692–7698.
145. T. Ikeda, T. Sasaki, and K. Ichimura. Photochemical switching of polarization in ferroelectric liquid-crystal films. Nature 1993, 361, 428–430.
146. H. G. Walton and H. J. Coles. Photomechanically induced phase transitions in ferroelectric liquid crystals. Ferroelectrics 1993, 147, 223–240.
147. A. Langhoff and F. Giesselmann. Kinetics of the photoferroelectric effect in chiral smectic-C liquid crystals studied by time-resolved measurements of spontaneous electric polarization and director tilt angle. Chem. Phys. Chem. 2002, 3, 424–432.
148. L. Komitov, C. Ruslim, and K. Ichimura. Light-induced changes of the spontaneous polarization in ferroelectric liquid crystal phase. J. Nonlinear Opt. Phys. Mater. 2000, 9, 151–156.
149. L. M. Blinov, M. V. Kozlovsky, M. Ozaki, and K. Yoshino. Photochromism of azo dyes and effect of liquid crystalline ordering on its efficiency and kinetics. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. C: Mol. Mater. 1996, 6, 6235–6252.
150. A. Langhoff and F. Giesselmann. Thermodynamics of the photoferroelectric effect in smectic-C* liquid crystals. J. Chem. Phys. 2002, 177, 2232–2237.
151. L. Dinescu, K. E. Maly, and R. P. Lemieux. Design of photonic liquid crystal materials: synthesis and evaluation of new chiral thioindigo dopants designed to photomodulate the spontaneous polarization of ferroelectric liquid crystals. J. Mater. Chem. 1999, 9, 1679–1686.
152. R. P. Lemieux. Photoswitching of ferroelectric liquid crystals using photochromic dopants. Soft Matter 2005, 1, 348–354.
153. K. Shirota and I. Yamaguchi. Optical switching of antiferroelectric liquid crystal with azo dye using photochemically induced SmCA*-SmC* phase transition. Jpn. J. Appl. Phys., Part 2 1997, 36, L1035–L1037.
154. H. J. Coles, H. G. Walton, D. Guillon, and G. Poetti. Photomechanically induced phase transitions in ferroelectric liquid crystals. Liq. Cryst. 1993, 15, 551–558.
155. R. K. Vijayaraghavan, S. Abraham, H. Akiyama, S. Furumi, N. Tamaoki, and S. Das. Photoresponsive glass-forming butadiene-based chiral liquid crystals with circularly polarized photoluminescence. Adv. Funct. Mater. 2008, 18, 2510–2517.
156. T. Matsui, K. Nakayama, M. Ozaki, and K. Yoshino. Photoinduced layer alignment control in ferroelectric liquid crystal with N*–C* phase transition doped with photochromic dye. Appl. Phys. Lett. 2000, 76, 1228–1230.
157. For a recent general review on discotic liquid crystals see: S. Kumar. Chemistry of Discotic Liquid Crystals: From Monomers to Polymers, CRC Press, Boca Raton, FL, 2010.
158. Y. Shimizu, A. Kurobe, H. Monobe, N. Terasawa, K. Kiyohara, and K. Uchida. Calamitic and discotic mesophases formed by kinetically controlled rod–disc alternation of molecular shape in a triphenylene–azobenzene mesogenic system. Chem. Commun. 2003, 1676–1677.
159. S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu, and A. Kitamura. Hierarchical organization of photoresponsive hydrogen-bonded rosettes. J. Am. Chem. Soc. 2005, 127, 11134–11139.
160. C. S. Pecinovsky, E. S. Hatakeyama, and D. L. Gin. Polymerizable photochromic macrocyclic metallomesogens: design of supramolecular polymers with responsive nanopores. Adv. Mater. 2008, 20, 174–178.
161. E. Westphal, I. H. Bechtold, and H. Gallardo. Synthesis and optical/thermal behavior of new azo photoisomerizable discotic liquid crystals. Macromolecules 2010, 43, 1319–1328.
162. Y. Norikane, Y. Hirai, and M. Yoshida. Photoinduced isothermal phase transitions of liquid-crystalline macrocyclic azobenzenes. Chem. Commun. 2011, 47, 1770–1772.
163. C. V. Lobo, S. K. Prasad, and C. V. Yelamaggad. Experimental investigations on weakly polar liquid crystal–aerosil composites. J. Phys.: Condens. Matter. 2006, 18, 767–776.
164. G. S. Iannacchione. Review of liquid-crystal phase transitions with quenched random disorder. Fluid Phase Equilib. 2004, 222–223, 177–187.
165. M. Caggioni, A. Roshi, S. Barjami, F. Mantegazza, G. S. Iannacchione, and T. Bellini. Isotropic to nematic transition of aerosil-disordered liquid crystals, Phys. Rev. Lett. 2004, 93, 127801-1–127801-4.
166. G. S. Iannacchione, C. W. Garland, J. T. Mang, and T. P. Rieker. Calorimetric and small angle X-ray scattering study of phase transitions in octylcyanobiphenyl-aerosil dispersions. Phys. Rev. E 1998, 58, 5966–5981.
167. V. Jayalakshmi, G. G. Nair, and S. K. Prasad. Effect of aerosil dispersions on the photoinduced nematic–isotropic transition. J. Phys.: Condens. Matter. 2007, 19, 226213-1–226213-12.
168. K. L. Sandhya, S. K. Prasad, and G. G. Nair. Enhanced dynamic response of the photoinduced nematic–isotropic transition in a polymer matrix. Appl. Phys. Lett. 2003, 83, 2707–2709.
169. K. Miyano. Wall-induced pretransitional birefringence: a new tool to study boundary aligning forces in liquid crystals. Phys. Rev. Lett. 1979, 43, 51–54.
170. H. Orendi and M. Ballauff. Complete phase diagrams of mixtures of a nematic liquid crystal with n-alkanes. Liq. Cryst. 1989, 6, 497–500.
171. G. G. Nair, S. K. Prasad, and G. Hegde. Influence of a long-chain alkane on the photoinduced nematic–isotropic transition. Phys. Rev. E 2004, 69, 021708-1–021708-6.
172. G. Hegde, S. K. Prasad, and G. G. Nair. Unpublished work.
173. V. Jayalakshmi, G. G. Nair, and S. K. Prasad. Kinetics of the thermal back relaxation time of the photoinduced nematic–isotropic transition. Phys. Rev. E 2007, 75, 031710-1–031710-6.
174. S. K. Prasad, G. G. Nair, and V. Jayalakshmi. Electric-field-assisted acceleration of the photostimulated nematic–isotropic transition. Adv. Mater. 2008, 20, 1363–1367.
175. S. K. Prasad, D. S. S. Rao, and P. Jeyagopal. Effect of pressure on the photoinduced nematic–isotropic phase transition. Phys. Rev. E 2001, 64, 011706-1–011706-4.
176. S. K. Prasad, V. K. Gupta, D. S. S. Rao, and C. V. Lobo. Effect of pressure on the dynamics of the photostimulated orientational ordering transition in a liquid crystal. Phys. Rev. E 2005, 72, 021705-1–021705-6.
177. V. K. Gupta, D. S. S. Rao, C. V. Lobo, S. K. Prasad, U. S. Hiremath, and C. V. Yelamaggad. High pressure investigations of the photo-stimulated orientational ordering transition in a liquid crystal with photoactive dimeric molecules. Thermochim. Acta 2006, 440, 205–211.
178. R. V. Tranfield and P. J. Collings. High-pressure measurements in a homologous series of liquid crystals. Phys. Rev. A 1982, 25, 2744–2749.
179. G. G. Nair, S. K. Prasad, and C. V. Yelamaggad. Opto-dielectric effect on a nematic liquid crystal doped with a photoactive azo mesogen. J. Appl. Phys. 2000, 87, 2084–2089.
180. S. Krishna Prasad, K. L. Sandhya, G. G. Nair, U. S. Hiremath, and C. V. Yelamaggad. Spacer parity dependence of photoinduced effects in liquid-crystalline dimers. J. Appl. Phys. 2002, 92, 838–841.
181. C. S. Hudson. Die gegenseitige loslichkeit von nikotin in wasser. Z. Physik. Chem. 1904, 47, 113–115.
182. For an exhaustive survey of reentrance in a variety of systems see: T. Narayanan and A. Kumar. Reentrant phase transitions in multicomponent liquid mixtures. Phys. Rep. 1994, 249, 135–218.
183. P. E. Cladis. New liquid-crystal phase diagram. Phys. Rev. Lett. 1975, 35, 48–51.
184. P. E. Cladis, R. K. Bogardus, W. B. Daniels, and G. N. Taylor. High-pressure investigation of the reentrant nematic bilayer smectic-A transition. Phys. Rev. Lett. 1977, 39, 720–723.
185. F. Hardouin, G. Sigaud, M. F. Achard, and H. Gasparoux. Enantiotropic nematic reentrant behaviour at atmospheric pressure in a pure compound. Phys. Lett. A 1979, 71, 347–349.
186. N. V. Madhusudhana, B. K. Sadashiva, and K. P. L. Moodithaya. Curr. Sci. 1979, 48, 613–614.
187. S. K. Prasad and G. G. Nair. Effects of photo-controlled nanophase segregation in a re-entrant nematic liquid crystal. Adv. Mater. 2001, 13, 40–43.
188. S. K. Prasad, G. G. Nair, and G. Hegde. Dynamic self-assembly of the liquid crystalline smectic A phase. Adv. Mater. 2005, 17, 2086–2091.
189. S. K. Prasad, G. G. Nair, and G. Hegde. Nonequilibrium liquid crystalline layered phase stabilized by light. J. Phys. Chem. B 2007, 111, 345–350.
190. Y. Lansac, M. A. Glaser, N. A. Clark, and O. D. Lavrentovich. Photocontrolled nanophase segregation in a liquid-crystal solvent. Nature 1999, 398, 54–57.
191. A. N. Berker and J. S. Walker. Frustrated spin-gas model for doubly reentrant liquid crystals. Phys. Rev. Lett. 1981, 47, 1469–1472.
192. X. Tong, M. Pelletier, A. Lasia, and Y. Zhao. Fast cis–trans isomerization of an azobenzene derivative in liquids and liquid crystals under a low electric field to the photoinduced N–I transition. Angew. Chem. Int. Ed. 2008, 47, 3596–3599.
193. S. Sridevi, S. K. Prasad, and G. G. Nair. Electric-field-dictated phase diagram and accelerated dynamics of a reentrant nematic liquid crystal under photostimulation. Phys. Rev. E 2009, 80, 021703-1–021703-9.
194. G. Fuchsel, T. Klamroth, J. Dokic, and P. Saalfrank. On the electronic structure of neutral and ionic azobenzenes and their possible role as surface mounted molecular switches. J. Phys. Chem. B 2006, 110, 16337–16345.
195. M. Alemani, F. Moresco, M. V. Peters, S. Hecht, K.-H. Rieder, and L. Grill. Electric field-induced isomerization of azobenzene by STM. J. Am. Chem. Soc. 2006, 128, 14446–14447.
196. A. D. L. Chandani, E. Gorecka, Y. Ouchi, H. Takezoe, and A. Fukuda. Antiferroelectric chiral smectic phases responsible for the trislable switching in MHPOBC. Jpn. J. Appl. Phys., C Part 2 1989, 28, L1265–L1268.
197. S. T. Lagerwall. Ferroelectric and Antiferroelectric Liquid Crystals, Wiley-VCH, Weinheim, 1999.
198. I. Musevic, R. Blinc, and B. Zeks. The Physics of Ferroelectric and Antiferroelectric Liquid Crystals, World Scientific, Singapore, 2000.
199. See e., g., P. Mach, R. Pindak, A. M. Levelut, P. Barois, H. T. Nguyen, C. C. Huang, and L. Furenlid. Structural characterization of various chiral smectic-C phases by resonant X-ray scattering. Phys. Rev. Lett. 1998, 81, 1015–1018.
200. S. K. Prasad, K. L. Sandhya, and Y. S. Negi. Photoinduced effects in the vicinity of the smectic-Cα*–smectic-A transition. Phys. Rev. E 2002, 65, 031718-1–031718-4.
201. S. K. Prasad, K. L. Sandhya, D. S. S. Rao, and Y. S. Negi. Time-resolved measurements of the dynamics of the photoinduced smectic-Cα*–smectic-A transition. Phys. Rev. E 2003, 67, 051701-1–051701-5.
202. S. K. Prasad, S. M. Khened, V. N. Raja, S. Chandrasekhar, and B. Shivakumar. Dielectric studies in the vicinity of the A-C* transition. Ferroelectrics 1993, 138, 37–49.
203. C. Bagnuls and C. Bervillier. Nonasymptotic critical behaviour from field theory at d = 3: the disordered-phase case. Phys. Rev. B 1985, 32, 7209–7231.
204. K. Ema, M. Ogawa, A. Takagi, and H. Yao. Crossover from XY critical to tricritical behaviour of heat capacity at the smectic-A–chiral-smectic-C liquid-crystal transition. Phys. Rev. E 1996, 54, R25–R28.
205. G. G. Nair, G. Hegde, S. K. Prasad, C. V. Lobo, and Y. S. Negi. Photoinduced effects in the vicinity of the smectic-A–smectic-CA* transition: polarization, tilt angle, and response time studies. Phys. Rev. E 2006, 73, 011712-1–011712-8.
206. T. Carlsson, B. Zeks, C. Filipic, A. Levstik, and R. Blinc. Thermodynamic model of ferroelectric chiral smectic C* liquid crystals. Mol. Cryst. Liq. Cryst. 1988, 163, 11–72.
207. G. G. Nair, G. Hegde, S. K. Prasad, and Y. S. Negi. Investigations of the opto-dielectric effects in the vicinity of the smectic-A–smectic-CA* transition. J. Phys.: Condens. Matter. 2006, 18, 9415–9425.
208. G. G. Nair, S. K. Prasad, U. S. Hiremath, and C. V. Yelamaggad. Effect of light on the polarization of a banana-shaped achiral compound doped with a photoactive azobenzene material. J. Appl. Phys. 2001, 90, 48–52.
209. M. Mathews, R. S. Zola, D. -ke Yang, and Q. Li. Thermally, photochemically and electrically switchable reflection colors from self-organized chiral bent-core liquid crystals. J. Mater. Chem. 2011, 21, 2098–2103.
210. S. Pieraccini, S. Masiero, A. Ferrarini, and G. P. Spada. Chirality transfer across length-scales in nematic liquid crystals: fundamentals and applications. Chem. Soc. Rev. 2011, 40, 258–271.
211. T. van Leeuwen, T. C. Pijper, J. Areephong, B. L. Feringa, W. R. Browne, and N. Katsonis. Reversible photochemical control of cholesteric liquid crystals with a diamine-based diarylethene chiroptical switch. J. Mater. Chem. 2011, 21, 3142–3146.
212. G. Hegde, G. G. Nair, S. K. Prasad, and C. V. Yelamaggad. A photodriven dual-frequency addressable optical device. J. Appl. Phys. 2005, 97, 093105-1–093105-6.
213. M. H. Li and P. Keller. Artificial muscles based on liquid crystal elastomers. Phil. Trans. R. Soc. A 2006, 364, 2763–2777.
214. D. K. Shenoy, D. L. Thomsen, A. Srinivasan, P. Keller, and B. R. Ratna. Carbon coated liquid crystal elastomer film for artificial muscle applications. Sens. Actuators A 2002, 96, 184–188.
215. H. Finkelmann, S. T. Kim, A. Munoz, P. Palffy-Muhoray, and B. Taheri. Tunable mirrorless lasing in cholesteric liquid crystalline elastomers. Adv. Mater. 2001, 13, 1069–1072.
216. H. S. Blair, H. I. Pague, and J. E. Riordan. Photoresponsive effects in azo polymers. Polymer 1980, 21, 1195–1198.
217. C. L. van Oosten, D. Corbett, D. Davies, M. Warner, C. W. M. Bastiaansen, and D. J. Broer. Bending dynamics and directionality reversal in liquid crystal network photoactuators. Macromolecules 2008, 41, 8592–8596.
218. M. Kondo, R. Miyasato, Y. Naka, J. Mamiya, M. Kinoshita, Y. Yu, C. J. Barrett, and T. Ikeda. Photomechanical properties of azobenzene liquid-crystalline elastomers. Liq. Cryst. 2009, 36, 1289–1293.
219. P. Beyer and R. Zentel. Photoswitchable smectic liquid-crystalline elastomers. Macromol. Rapid Commun. 2005, 26, 874–879.