
3.3. Explosion Limits and Oxidation Characteristics of Hydrogen
Many of the early contributions to the understanding of hydrogen–oxygen oxidation mechanisms developed from the study of explosion limits. Many extensive treatises were written on the subject of the hydrogen–oxygen reaction and, in particular, much attention was given to the effect of walls on radical destruction (a chain termination step) [2]. Such effects are not important in the combustion processes of most interest here; however, Appendix C details a complex modern mechanism based on earlier thorough reviews [3,4].
Flames of hydrogen in air or oxygen exhibit little or no visible radiation, what radiation one normally observes being due to trace impurities. Considerable amounts of OH can be detected, however, in the ultraviolet region of the spectrum. In stoichiometric flames, the maximum temperature reached in air is about 2400 K and in oxygen about 3100 K. The burned gas composition in air shows about 95–97% conversion to water, the radicals H, O, and OH comprising about one-quarter of the remainder [5]. In static systems practically no reactions occur below 675 K, and above 850 K explosion occurs spontaneously in the moderate pressure ranges. At very high pressures the explosion condition is moderated owing to a third-order chain terminating reaction, reaction (3.5), as will be explained in the following paragraphs.
It is now important to stress the following points in order to eliminate possible confusion with previously held concepts and certain subjects to be discussed later. The explosive limits are not flammability limits. Explosion limits are the pressure–temperature boundaries for a specific fuel–oxidizer mixture ratio that separates the regions of slow and fast reaction. For a given temperature and pressure, flammability limits specify the lean and rich fuel–oxidizer mixture ratio beyond which no flame will propagate. Next, recall that one must have fast reactions for a flame to propagate. A stoichiometric mixture of H2 and O2 at standard conditions will support a flame because an ignition source initially brings a local mixture into the explosive regime, whereupon the established flame by diffusion heats fresh mixture to temperatures high enough to be explosive. Thus, in the early stages of any flame, the fuel–air mixture may follow a low-temperature steady reaction system and in the later stages, an explosive reaction system. This point is significant, especially in hydrocarbon combustion, because it is in the low-temperature regime that particular pollutant-causing compounds are formed.
Figure 3.2 depicts the explosion limits of a stoichiometric mixture of hydrogen and oxygen. Explosion limits can be found for many different mixture ratios. The point X on Figure 3.2 marks the conditions (773 K; 1 atm) described at the very beginning of this chapter in Figure 3.1. It now becomes obvious that either increasing or decreasing the pressure at constant temperature can cause an explosion.
Certain general characteristics of this curve can be stated. First, the third limit portion of the curve is as one would expect from simple density considerations. Next, the first, or lower, limit reflects the wall effect and its role in chain destruction. For example, HO2 radicals combine on surfaces to form H2O and O2. Note the expression developed for αcrit (Eqn (3.11)) applies to the lower limit only when the wall effect is considered as a first-order reaction of chain destruction, since  destruction was written. Although the features of the movement of the boundaries are not explained fully, the general shape of the three limits can be explained by reasonable hypotheses of mechanisms. The manner in which the reaction is initiated to give the boundary designated by the curve in Figure 3.2 suggests, as was implied earlier, that the explosion is in itself a branched chain phenomenon. Thus, one must consider possible branched chain mechanisms to explain the limits.
destruction was written. Although the features of the movement of the boundaries are not explained fully, the general shape of the three limits can be explained by reasonable hypotheses of mechanisms. The manner in which the reaction is initiated to give the boundary designated by the curve in Figure 3.2 suggests, as was implied earlier, that the explosion is in itself a branched chain phenomenon. Thus, one must consider possible branched chain mechanisms to explain the limits.

Basically, only thermal, not photolytic, mechanisms are considered. The dissociation energy of hydrogen is less than that of oxygen, so the initiation can be related to hydrogen dissociation. Only a few radicals are required to initiate the explosion in the region of temperature of interest, that is, about 675 K. If hydrogen dissociation is the chain's initiating step, it proceeds by the reaction
![]() (3.18)
(3.18)
which requires about 435 kJ/mol.
The early modeling literature suggested the initiation step
 (3.19)
(3.19)
because this reaction requires only 210 kJ/mol, but this trimolecular reaction has been evaluated to have only a very slow rate [6]. Because in modeling it accurately reproduces experimental ignition delay measurements under shock tube and detonation conditions [7], the most probable initiation step, except at the very highest temperature at which reaction (3.18) would prevail, could be
![]() (3.20)
(3.20)
where HO2 is the relatively stable hydroperoxy radical, which has been identified by mass spectroscopic analysis. There are data now that support this initiation reaction in the temperature range 1662–2097 K [8].
The essential feature of the initiation step is to provide a radical for the chain system and, as discussed in the previous section, the actual initiation step is not important in determining the explosive condition, nor is it important in determining the products formed. Either reaction (3.18) or (3.20) provides an H radical that develops a radical pool of OH, O, and H by the chain reactions
![]() (3.21)
(3.21)
![]() (3.22)
(3.22)
![]() (3.23)
(3.23)
![]() (3.24)
(3.24)
Reaction (3.21) is chain branching and 66 kJ/mol endothermic. Reaction (3.22) is also chain branching and 8 kJ/mol exothermic. Note that the H radical is regenerated in the chain system and there is no chemical mechanism barrier to prevent the system from becoming explosive. Since radicals react rapidly, their concentration levels in many systems are very small; consequently, the reverse of reactions (3.21), (3.22) and (3.24) can be neglected during the initiation process and early reactant consumption. Normally, reactions between radicals are not considered, except in termination steps late in the reaction when the concentrations are high and only stable product species exist. Thus, the reverse reactions (3.21), (3.22) and (3.24) are not important for the determination of the second limit, nor are they important for the steady-slow H2–O2 and CO–H2O–O2 reactions. However, they are generally important in all explosive H2–O2 and CO–H2O–O2 reactions. The importance of these radical–radical reactions in these cases is verified by the existence of superequilibrium radical concentrations and the validity of the partial equilibrium assumption.
The sequence (Eqns (3.21)–(3.24)) is of great importance in the oxidation reaction mechanisms of any hydrocarbon in that it provides the essential chain branching and propagating steps as well as the radical pool for fast reaction.
The important chain termination steps in the static explosion experiments (Figure 3.1) are
![]()
![]()
Either or both of these steps explain the lower limit of explosion, since it is apparent that wall collisions become much more predominant at lower pressure than molecular collisions. The fact that the limit is found experimentally to be a function of the containing vessel diameter is further evidence of this type of wall destruction step.
The second explosion limit must be explained by gas phase production and destruction of radicals. This limit is found to be independent of vessel diameter. For it to exist, the most effective chain branching reaction (reaction (3.21)) must be overridden by another reaction step. When a system at a fixed temperature moves from a lower to higher pressure, the system goes from an explosive to a steady reaction condition, so the reaction step that overrides the chain branching step must be more pressure-sensitive. This reasoning leads one to propose a third-order reaction in which the species involved are in large concentration [2]. The accepted reaction that satisfies these prerequisites is
![]() (3.25)
(3.25)
where M is the usual third body that takes away the energy necessary to stabilize the combination of H and O2. At higher pressures, it is certainly possible to obtain proportionally more of this trimolecular reaction than the binary system represented by reaction (3.21). The hydroperoxy radical HO2 is considered to be relatively unreactive so that it is able to diffuse to the wall and thus become a means for effectively destroying H radicals.
The upper (third) explosion limit is due to a reaction that overtakes the stability of the HO2 and is possibly the sequence
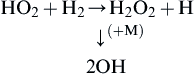 (3.26)
(3.26)
The reactivity of HO2 is much lower than that of OH, H, or O; therefore, somewhat higher temperatures are necessary for sequence (Eqn (3.26)) to become effective [9]. Water vapor tends to inhibit explosion due to the effect of reaction (3.25) in that H2O has a high third-body efficiency, which is most probably due to some resonance energy exchange with the HO2 formed.
Since reaction (3.25) is a recombination step requiring a third body, its rate decreases with increasing temperature, whereas the rate of reaction (3.21) increases with temperature. One then can generally conclude that reaction (3.21) will dominate at higher temperatures and lower pressures, while reaction (3.25) will be more effective at higher pressures and lower temperatures. Thus, in order to explain the limits in Figure 3.2, it becomes apparent that at temperatures above 875 K, reaction (3.21) always prevails, and the mixture is explosive for the complete pressure range covered.
In this higher-temperature regime and in atmospheric-pressure flames, the eventual fate of the radicals formed is dictated by recombination. The principal gas phase termination steps are
![]() (3.27)
(3.27)
![]() (3.28)
(3.28)
![]() (3.29)
(3.29)
![]() (3.30)
(3.30)
In combustion systems other than those whose lower-temperature explosion characteristics are represented in Figure 3.2, there are usually ranges of temperature and pressure in which the rates of reactions (3.21) and (3.25) are comparable. This condition can be specified by the simple ratio
![]()
Indeed, in developing complete mechanisms for the oxidation of CO and hydrocarbons applicable to practical systems over a wide range of temperatures and high pressures, it is important to examine the effect of the HO2 reactions when the ratio is as high as 10 or as low as 0.1. Considering that for air combustion the total concentration (M) can be that of nitrogen, the boundaries of this ratio are depicted in Figure 3.3, as derived from the data in Appendix C. These reaction rate data indicate that the second explosion limit, as determined by glass vessel experiments and many other experimental configurations, as shown in Figure 3.2, has been extended (Figure 3.4) and verified experimentally [9]. Thus, to be complete for the H2–O2 system and other oxidation systems containing hydrogen species, one must also consider reactions of HO2. Sometimes HO2 is called a metastable species because it is relatively unreactive as a radical. Its concentrations can build up in a reacting system. Thus, HO2 may be consumed in the H2–O2 system by various radicals according to the following reactions [4]:
![]() (3.31)
(3.31)
![]() (3.32)
(3.32)
![]() (3.33)
(3.33)
![]() (3.34)
(3.34)
![]() (3.35)
(3.35)


The recombination of HO2 radicals by
![]() (3.36)
(3.36)
yields hydrogen peroxide (H2O2), which is consumed by reactions with radicals and by thermal decomposition according to the following sequence:
![]() (3.37)
(3.37)
![]() (3.38)
(3.38)
![]() (3.39)
(3.39)
![]() (3.40)
(3.40)
From the sequence of reactions (3.37)–(3.40), one finds that although reaction (3.25) terminates the chain under some conditions, under other conditions it is part of a chain propagating path consisting essentially of reactions (3.25) and (3.32) or reactions (3.25), (3.36) and (3.40). It is also interesting to note that, as are most HO2 reactions, these two sequences of reactions are very exothermic; that is,

and

Hence, they can significantly affect the temperature of a (adiabatic) system and thereby move the system into an explosive regime. The point to be emphasized is that slow competing reactions can become important if they are very exothermic.
It is apparent that the fate of the H atom (radical) is crucial in determining the rate of the H2–O2 reaction or, for that matter, the rate of any hydrocarbon oxidation mechanism. From the data in Appendix C, one observes that at temperatures encountered in flames the rates of reaction between H atoms and many hydrocarbon species are considerably larger than the rate of the chain branching reaction (3.21). Note the comparisons in Table 3.1. Thus, these reactions compete very effectively with reaction (3.21) for H atoms and reduce the chain branching rate. For this reason, hydrocarbons act as inhibitors for the H2–O2 system [4]. As implied, at highly elevated pressures (P ≥ 20 atm) and relatively low temperatures (T ≅ 1000 K), reaction (3.25) will dominate over reaction (3.21), and as shown, the sequence of reactions (3.25), (3.36) and (3.40) provides the chain propagation. Also, at higher temperatures, when H + O2 → OH + O is microscopically balanced, reaction (3.25) (H + O2 + M → HO2 + M) can compete favorably with reaction (3.21) for H atoms, since the net removal of H atoms from the system by reaction (3.21) may be small due to its equilibration. In contrast, when reaction (3.25) is followed by the reaction of the fuel with HO2 to form a radical and hydrogen peroxide and then by reaction (3.40), the result is chain branching. Therefore, under these conditions, increased fuel will accelerate the overall rate of reaction and will act as an inhibitor at lower pressures due to competition with reaction (3.21) [4].
The detailed rate constants for all the reactions discussed in this section are given in Appendix C. The complete mechanism for CO or any hydrocarbon or hydrogen-containing species should contain the appropriate reactions of the H2–O2 steps listed in Appendix C; one can ignore the reactions containing Ar as a collision partner in real systems. It is important to understand that, depending on the temperature and pressure of concern, one need not necessarily include all the H2–O2 reactions. It should be realized as well that each of these reactions is a set comprising a forward and a backward reaction; but, as the reactions are written, many of the backward reactions can be ignored. Recall that the backward rate constant can be determined from the forward rate constant and the equilibrium constant for the reaction system.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.