
Chapter 6. Fires and Explosions
The learning objectives for this chapter are to:
Describe the nature of fires and explosions.
Define the fire triangle and explain how to use it to prevent flammable mixtures.
Characterize the flammability of gases, liquids, and dusts.
Estimate flammability parameters for mixtures.
Draw a flammability triangle diagram and apply it.
Describe explosions and how to estimate damage.
Chemicals present a substantial hazard in the form of fires and explosions. The combustion of a few liters of toluene can destroy an ordinary chemistry laboratory in minutes; persons present may be killed. The potential consequences of fires and explosions in pilot plants and plant environments are even greater.
Chemical and hydrocarbon plant losses resulting from fires and explosions are substantial. In the 100 largest incidents in the chemical industry, fires and explosions accounted for $25.5 billion in damage and represented the largest fraction of all losses.1 These losses include only property damage; they exclude business interruption or other losses.
1The 100 Largest Losses 1974–2015 (New York, NY: J. H. Marsh & McLennan, 2016).
To prevent accidents resulting from fires and explosions, engineers must be familiar with
The fire and explosion properties of materials,
The nature of the fire and explosion process, and
Procedures to eliminate, reduce, or control fire and explosion hazards.
This chapter covers the first two topics, emphasizing definitions and calculation methods for estimating the magnitude and consequences of fires and explosions. Procedures to reduce fire and explosion hazards are presented in Chapter 7.
6-1 The Fire Triangle
The three essential elements for combustion are a fuel, an oxidizer, and an ignition source. These elements are illustrated by the fire triangle, shown in Figure 6-1.

Some experts add one or more legs to the fire triangle, such as fuel in sufficient quantity or an ignition source of adequate energy. For all practical purposes and for application in a plant environment, however, the fire triangle is sufficient and easy to both understand and communicate.
Fire, or burning, is the rapid exothermic oxidation of an ignited fuel. The fuel can be in solid, liquid, or vapor form, although vapor and liquid fuels are generally easier to ignite. The combustion always occurs in the vapor phase: liquids are volatized and solids are decomposed into vapor before combustion.
When a fuel, an oxidizer, and an ignition source are present at the necessary levels, burning will occur. Conversely, a fire will not occur if (1) a fuel is not present or is not present in sufficient quantities, (2) an oxidizer is not present or is not present in sufficient quantities, and (3) the ignition source is not energetic enough to initiate the fire.
Two common examples of the three components of the fire triangle are (1) wood, air, and a match and (2) gasoline, air, and a spark. However, other, less obvious combinations of chemicals can lead to fires and explosions. Indeed, various fuels, oxidizers, and ignition sources are commonly encountered in the chemical industry:
Fuels
Liquids: gasoline, acetone, ether, pentane
Solids: plastics, wood dust, fibers, metal particles
Gases: natural gas, acetylene, propane, carbon monoxide, hydrogen
Oxidizers
Gases: oxygen, fluorine, chlorine
Liquids: hydrogen peroxide, nitric acid, perchloric acid
Solids: metal peroxides, ammonium nitrite
Ignition Sources
Sparks, flames, static electricity, heat
In the past, the sole method for controlling fires and explosions was elimination of or reduction in ignition sources. Practical experience has shown that this approach is not robust enough: The ignition energies for most flammable materials are too low and ignition sources too plentiful. As a result, the primary means to prevent fires and explosion is to prevent flammable mixtures, while continuing to eliminate ignition sources.
6-2 Distinction between Fires and Explosions
The major distinction between fires and explosions is the rate of energy release. Fires release energy slowly, whereas explosions release energy rapidly, typically on the order of microseconds to tens of milliseconds. Fires can also result from explosions, and explosions can result from fires.
A good example of how the energy release rate affects the consequences of an accident is a standard automobile tire. The compressed air within the tire contains energy. If the energy is released slowly through the nozzle, the tire is harmlessly deflated. In contrast, if the tire ruptures suddenly and all the energy within the compressed tire releases rapidly, the result is a dangerous explosion.
6-3 Definitions
Some of the commonly used definitions related to fires and explosions are given here. These definitions are discussed in greater detail in later sections.
Combustion or fire: Combustion or fire is a chemical reaction in which a substance combines with an oxidant and releases energy. Part of the energy released is used to sustain the reaction.
Ignition: Ignition may be caused by a flammable mixture coming in contact with a source of ignition with sufficient energy or the gas reaching a temperature high enough to cause the gas to auto-ignite.
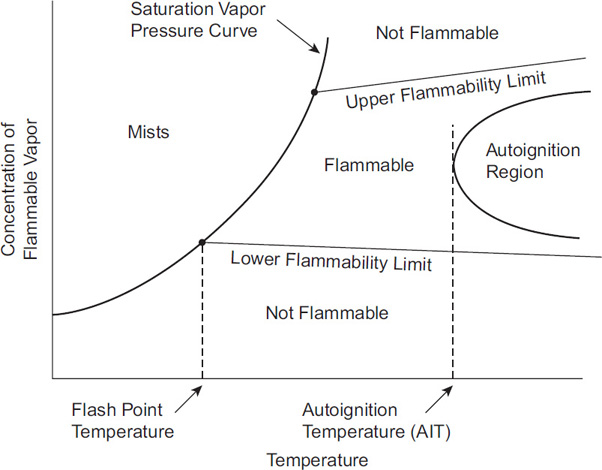
Autoignition temperature (AIT): The AIT is a fixed temperature above which adequate energy is available in the environment to provide an ignition source—that is, an explicit ignition source is not required.
Flash point temperature: The flash point temperature of a liquid is the lowest temperature at which it gives off enough vapor to form an ignitable mixture with air. At the flash point, the vapor will usually burn only briefly, because inadequate vapor is produced to maintain combustion. The flash point temperature generally increases with increasing pressure.
Fire point: The fire point is the lowest temperature at which a vapor above a liquid will continue to burn once ignited. This temperature is higher than the flash point.
Flammability limits: Flammability limits are only defined in air at room temperature and pressure; they have units of volume percent fuel in air. Combustion of an air–fuel mixture will occur only between the lower flammable limit (LFL) and the upper flammable limit (UFL). Below the LFL, the mixture is too lean to burn. Above the UFL, the mixture is too rich in fuel. Lower explosion limit (LEL) and upper explosion limit (UEL) are used interchangeably with LFL and UFL, respectively.
Explosion: An explosion is a rapid expansion of gases resulting in a rapidly moving pressure or shock wave. The expansion can be mechanical (by means of a sudden rupture of a pressurized vessel), or it can be the result of a rapid chemical reaction. Explosion damage is caused by the pressure or shock wave.
Mechanical explosion: This type of explosion results from the sudden failure of a vessel containing high-pressure nonreactive gas.
Deflagration: In this type of explosion, the reaction front moves at a speed less than the speed of sound in the unreacted medium.
Detonation: In this type of explosion, the reaction front moves at a speed greater than the speed of sound in the unreacted medium.
Confined explosion: This type of explosion occurs within a vessel, process volume, or building.
Unconfined explosion: Unconfined explosions occur in the open and are usually the result of a flammable gas release. The gas is dispersed and mixed with air until it comes in contact with an ignition source. Unconfined explosions are rarer than confined explosions because the explosive material is frequently diluted below the LFL by wind dispersion. These explosions are very destructive because large quantities of flammable gas and large areas are frequently involved.
Boiling-liquid expanding-vapor explosion (BLEVE): A BLEVE occurs due to the rupture of a vessel that contains a liquid at a temperature above its atmospheric pressure boiling point. The subsequent BLEVE is the explosive vaporization of a large fraction of the vessel contents, possibly followed by combustion or explosion of the vaporized cloud if it is combustible. This type of explosion can occur when an external fire heats the contents of a tank of volatile material. As the temperature of the tank contents increases, the vapor pressure of the liquid within the tank increases and the tank’s structural integrity is reduced. If the tank ruptures, the hot liquid volatilizes explosively.
Dust explosion: This type of explosion results from the rapid combustion of fine solid particles. Many solid materials (including common metals such as iron and aluminum) become flammable and explosive when reduced to a fine powder.
Shock wave: A shock wave is an abrupt or nearly instantaneous pressure wave moving through a gas. A shock wave in open air is followed by a strong wind; this combination of shock wave and wind is called a blast wave. The pressure increase in the shock wave is so rapid that the process is mostly adiabatic.
Overpressure: This pressure on an object occurs as a result of an impacting shock wave.
Flammability parameters are not fundamental properties like heat capacity or density, but rather are highly dependent on the experimental apparatus and procedure. Thus, these parameters are not an absolute boundary between safe and unsafe operation. Care must be taken in applying these flammability parameters to practical situations.
Figure 6-2 is a plot of flammable vapor concentration versus temperature and shows how several of the preceding definitions are related. The exponential curve in the figure represents the saturation vapor pressure curve for the liquid material. Typically, the UFL increases and the LFL decreases with temperature. The LFL theoretically intersects the saturation vapor pressure curve at the flash point temperature, although experimental data do not always agree with this supposition. The autoignition temperature is actually the lowest temperature of an autoignition region. The behavior of the autoignition region and the flammability limits at higher temperatures are not known.
6-4 Flammability Characteristics of Liquids and Vapors
Flammability characteristics of some important organic chemicals (liquids and gases) are provided in Appendix B.
Liquids
The flash point temperature is the primary flammability parameter used to characterize the fire and explosion hazard of liquids.
Several different experimental methods are used to determine flash points, each of which produces a somewhat different value. The two most commonly used methods are open cup and closed cup, depending on the physical configuration of the experimental equipment. The open-cup flash point is typically a few degrees higher than the closed-cup flash point.
One method to determine the flash point temperature is by an open-cup apparatus, like that shown in Figure 6-3. The liquid to be tested is placed in the open cup. The liquid temperature is measured with a thermometer while a Bunsen burner is used to heat the liquid. A small flame is established on the end of a movable wand. During heating, the wand is slowly moved back and forth over the open liquid pool. Eventually a temperature is reached at which the liquid is volatile enough to produce a flammable vapor, and a momentary flashing flame occurs in the open cup. The temperature at which this first occurs is called the flash point temperature. Note that at the flash point temperature, only a momentary flame is expected. A higher temperature, called the fire point temperature, is required to produce a continuous flame.

The problem with the open-cup flash point apparatus is that air movements over the open cup may change the vapor concentrations and increase the experimentally determined flash point. To prevent this interference, most modern flash point methods employ a closed-cup procedure. In this apparatus, a small, manually opened shutter is provided at the top of the cup. The liquid is placed in a preheated cup and allowed to sit for a fixed time period. The shutter is then opened and the liquid is exposed to the flame. Closed-cup methods typically result in lower flash points.
The open-cup method shown in Figure 6-3 is frequently used to obtain an initial estimate of the flash point temperature since the temperature can be increased continuously in one experiment until a flash is obtained. Then, a closed-cup method is used to obtain a more precise value, since multiple trial-and-error runs must be done.
Satyanarayana and Rao showed that the flash point temperatures for pure materials correlate well with the boiling point of the liquid.2 They were able to fit the flash point for more than 1200 compounds with an error of less than 1% of the absolute temperature using the equation
2K. Satyanarayana and P. G. Rao. “Improved Equation to Estimate Flash Points of Organic Compounds.” Journal of Hazardous Materials 32 (1992): 81–85.
Tf=a+b(c/Tb)2 e−c/Tb(1−e−c/Tb)2(6-1)
where
Tf is the flash point temperature (K),
a, b, and c are constants provided in Table 6-1 (K), and
Table 6-1 Constants Used in Equation 6-1 for Predicting the Flash Point
Chemical group |
a |
b |
c |
|---|---|---|---|
Hydrocarbons |
225.1 |
537.6 |
2217 |
Alcohols |
230.8 |
390.5 |
1780 |
Amines |
222.4 |
416.6 |
1900 |
Acids |
323.2 |
600.1 |
2970 |
Ethers |
275.9 |
700.0 |
2879 |
Sulfur |
238.0 |
577.9 |
2297 |
Esters |
260.8 |
449.2 |
2217 |
Ketones |
260.5 |
296.0 |
1908 |
Halogens |
262.1 |
414.0 |
2154 |
Aldehydes |
264.5 |
293.0 |
1970 |
Phosphorus-containing |
201.7 |
416.1 |
1666 |
Nitrogen-containing |
185.7 |
432.0 |
1645 |
Petroleum fractions |
237.9 |
334.4 |
1807 |
Source: K. Satyanarayana and P. G. Rao. “Improved Equation to Estimate Flash Points of Organic Compounds.” Journal of Hazardous Materials 32 (1992): 81–85.
Tb is the boiling point temperature of the material (K).
Table 6-1 provides constants for Equation 6-1.
Flash points can be estimated for multicomponent mixtures if only one component is flammable and if the flash point temperature of the pure flammable component is known. In this case, the flash point temperature is estimated by determining the temperature at which the vapor pressure of the flammable component in the mixture is equal to the pure component vapor pressure at its flash point. Experimentally determined flash points are recommended for multicomponent mixtures with more than one flammable component.
Example 6-1
Methanol has a flash point of 54°F, and its vapor pressure at this temperature is 62 mm Hg. What is the flash point of a solution containing 75% methanol and 25% water by weight?
Solution
The mole fractions of each component are needed to apply Raoult’s law. Assuming a basis of 100 kg of solution, we obtain the following:
|
Kilograms |
Molecular weight |
Kg-moles |
Mole fraction |
|---|---|---|---|---|
Water |
25 |
18 |
1.39 |
0.37 |
Methanol |
75 |
32 |
2.34 |
0.63 |
3.73 |
1.00 |
Raoult’s law is used to compute the partial vapor pressure, P, of the methanol in the vapor, based on the saturation vapor pressure, Psat:
P=xPsatPsat=p/x=62/0.63=98.4 mm Hg
Using a graph of the vapor pressure versus temperature, shown in Figure 6-4, the flash point of the solution is 20.5°C (68.9°F).

Gas and Vapor Mixtures
LFLs and UFLs for gas and vapor mixtures are often needed. These mixture limits are computed using Le Châtelier’s equation:3
3H. Le Châtelier. “Estimation of Firedamp by Flammability Limits.” Annals of Mines 8, no. 19 (1891): 388–395.
LFLmix=1nΣt−1yiLFLi(6-2)
where
LFLi is the lower flammable limit for component i (in volume percent) of component i in fuel and air,
yi is the mole fraction of component i on a combustible basis, and
n is the number of combustible species.
Similarly, for the upper flammability limit,
UFLmix=1nΣt−1yiUFLi(6-3)
where UFLi is the upper flammable limit for component i (in volume percent) of component i in fuel and air.
Le Châtelier’s equation is empirically derived and is not universally applicable. Mashuga and Crowl derived Le Châtelier’s equation using thermodynamics.4 The derivation shows that the following assumptions are inherent in this equation:
4C. V. Mashuga and D. A. Crowl. “Derivation of Le Châtelier’s Mixing Rule for Flammable Limits.” Process Safety Progress 19, no. 2 (2000): 112–117.
The product heat capacities are constant.
The number of moles of gas is constant.
The combustion kinetics of the pure species is independent and unchanged by the presence of other combustible species.
The adiabatic temperature rise at the flammability limit is the same for all species.
These assumptions were found to be reasonably valid at the LFL and less so at the UFL.
Proper usage of Le Châtelier’s rule requires flammability limit data at the same temperature and pressure. Also, the flammability data reported in the literature may be from different sources, with wide variability in the data. Combining data from these different sources may cause unsatisfactory results, which may not be obvious to the user.
Example 6-2
What are the LFL and UFL of a gas mixture composed of 0.8% hexane, 2.0% methane, and 0.5% ethylene by volume?
Solution
The mole fractions on a fuel-only basis are calculated in the following table. The LFL and UFL data are obtained from Appendix B.
|
Volume % |
Mole fraction on combustible basis |
LFLi (volume %) |
UFLi (volume %) |
|---|---|---|---|---|
Hexane |
0.8 |
0.24 |
1.2 |
7.5 |
Methane |
2.0 |
0.61 |
5.0 |
15 |
Ethylene |
0.5 |
0.15 |
2.7 |
36.0 |
Total combustibles |
3.3 |
|
|
|
Air |
96.7 |
|
|
|
Equation 6-2 is used to determine the LFL of the mixture:
LFLmix=1n∑i=1yiLFLi=10.241.2+0.615.0+0.152.7=1/0.378=2.65% by volume combustibles
Equation 6-3 is used to determine the UFL of the mixture:
UFLmix=1n∑i=1yiUFLi=10.247.5+0.6115+0.1536.0=13.0 % by volume combustibles
Because the mixture contains 3.3% combustibles, it is flammable.
Flammability Limit Dependence on Temperature
In general, the flammability range increases with temperature.5 The following empirically derived equations are available for vapors:
5M. G. Zabetakis, S. Lambiris, and G. S. Scott. “Flame Temperatures of Limit Mixtures.” In Seventh Symposium on Combustion (London, UK: Butterworths, 1959), p. 484.
LFLT=LFL25−0.75ΔHc(T−25)(6-4)
LFLT=UFL25+0.75ΔHc(T−25)(6-5)
where
∆Hc is the net positive heat of combustion (kcal/mol), and
T is the temperature (°C).
Equations 6-4 and 6-5 are very approximate and work for only a very limited number of hydrocarbons over a limited temperature range. The 0.75 is equal to 100 Cp, with the heat capacity dominated by nitrogen.
Flammability Limit Dependence on Pressure
Pressure has little effect on the LFL except at very low pressures (less than 50 mm Hg absolute), where flames do not propagate.
The UFL increases significantly as the pressure is increased, broadening the flammability range. An empirical expression for the UFL for vapors as a function of pressure is available:6
UFLp=UFL+20.6(logP+1)(6-6)
6M. G. Zabetakis. “Fire and Explosion Hazards at Temperature and Pressure Extremes.” AICHE Symp., Vol. 2, Bureau of Mines, Pittsburgh, PA (1965).
where
P is the pressure (megapascals absolute) and
UFL is the upper flammable limit (volume percent fuel plus air at 1 atm).
Example 6-3
If the UFL for a substance is 11.0% by volume at 0.0 MPa gauge, what is the UFL at 6.2 MPa gauge?
Solution
The absolute pressure is P = 6.2 + 0.101= 6.301MPa.
The UFL is determined using Equation 6-6:
UFLP=UFL + 20.6 (log P+1)=11.0 + 20.6 (log 6.301 +1)=48 vol.% fuel in air.
This is a significant increase in the UFL and may be beyond the estimating capability of Equation 6-6.
Estimating Flammability Limits
For some situations, it may be necessary to estimate the flammability limits without experimental data. Flammability limits are easily measured; experimental determination is always recommended.
Jones found that for many hydrocarbon vapors the LFL and the UFL are a function of the stoichiometric concentration (Cst) of fuel:7
7G. W. Jones. “Inflammation Limits and Their Practical Application in Hazardous Industrial Operations.” Chemical Reviews 22, no. 1 (1938): 1–26.
LFL=0.55Cst(6-7)
UFL=3.50Cst(6-8)
where Cst is volume percent fuel in fuel plus air.
The stoichiometric concentration for most organic compounds is determined using the general combustion reaction
CmHxOy + z O2→mCO2+x2H2O(6-9)
It follows from the stoichiometry that
z=m+x4−y2
where z has units of moles O2/mole fuel.
Additional stoichiometric and unit changes are required to determine Cst as a function of z:
Cst=moles fuelmoles fuel + moles air×100=1001+(moles airmoles fuel)=1001+(10.21)(moles O2moles fuel)=1001+(z0.21)
Substituting z and applying Equations 6-7 and 6-8 yields
LFL=0.55(100)4.76m+1.19x−2.38y+1(6-10)
UFL=3.50(100)4.76m+1.19x−2.38y+1(6-11)
Another method correlates the flammability limits as a function of the heat of combustion of the fuel.8,9 A good fit was obtained for 123 organic materials containing carbon, hydrogen, oxygen, nitrogen, and sulfur. The resulting correlations are
8T. Suzuki. “Empirical Relationship between Lower Flammability Limits and Standard Enthalpies of Combustion of Organic Compounds.” Fire and Materials 18 (1994): 333–336.
9T. Suzuki and K. Koide. “Correlation between Upper Flammability Limits and Thermochemical Properties of Organic Compounds.” Fire and Materials 18 (1994): 393–397.
LFL=−−3.42ΔHc+0.569ΔHc+0.0538ΔH2c+1.80(6-12)
UFL=6.30ΔHc+0.567ΔH2c+23.5(6-13)
where
LFL and UFL are the lower and upper flammable limits (volume percent fuel in air), respectively, and
∆Hc is the positive heat of combustion for the fuel (in 103 kJ/mol).
Equation 6-13 is applicable only over the UFL range of 4.9–23%. If the heat of combustion is provided in kcal/mol, it can be converted to kJ/mol by multiplying by 4.184.
The prediction capability of Equations 6-6 through 6-13 is only modest at best. For hydrogen, the predictions are poor. For methane and the higher hydrocarbons, the results are improved. Thus, these methods should be used only for a quick initial estimate and should not replace actual experimental data.
Example 6-4
Estimate the LFL and the UFL for hexane, and compare the calculated limits to the actual values determined experimentally.
Solution
The stoichiometry is
C6H14+zO2→mCO2+x2H2O
and z, m, x, and y are found by balancing this chemical reaction using the definitions in Equation 6-9:
m=6 x=14 y=0 z=6+14/4-0=9.5
The LFL and the UFL are determined by using Equations 6-10 and 6-11:
LFL= 0.55(100) /[4.76(6)+1.19(14)+1]=1.19 vol. % versus 1.2 vol. % actualUFL=3.5(100) /[4.76(6)+1.19(14)+1]=7.57 vol. % versus 7.5 vol. % actual
Flammability limits, in general, are defined in air. As you will see later, flammable limits in pure oxygen are frequently useful for designing systems to prevent fires and explosions. Combustion in pure oxygen also exhibits a lower oxygen limit (LOL) and an upper oxygen limit (UOL), just like the LFL and UFL in air. These flammability limits have units of percent fuel in oxygen. Table 6-2 presents flammability data for a variety of fuels in pure oxygen. In general, for most common hydrocarbons, the LOL is close to the LFL.
Table 6-2 Flammability Limits in Pure Oxygen
|
|
Limits of flammability in pure oxygen |
|
|---|---|---|---|
Compound |
Formula |
Lower (LOL) |
Upper (UOL) |
Hydrogen |
H2 |
4.0 |
94 |
Deuterium |
D2 |
5.0 |
95 |
Carbon monoxidea |
CO |
15.5 |
94 |
Ammonia |
NH3 |
15.0 |
79 |
Methane |
CH4 |
5.1 |
61 |
Ethane |
C2H6 |
3.0 |
66 |
Ethylene |
C2H4 |
3.0 |
80 |
Propylene |
C3H6 |
2.1 |
53 |
Cyclopropane |
C3H6 |
2.5 |
60 |
Diethyl ether |
C4H10O |
2.0 |
82 |
Divinyl ether |
C4H6O |
1.8 |
85 |
aThe limits are insensitive to PH2o above a few mm Hg.
Source: Data from B. Lewis and G. von Elbe. Combustion, Flames, and Explosions of Gases (New York, NY: Harcourt Brace Jovanovich, 1987).
Hansen and Crowl derived an empirical equation for the UOL based on drawing lines along the flammable boundaries.10 They found that a good estimate of the UOL can be found from
UOL=UFL[100−CUOL(100−UFL0)]UFL0+UFL(1−CUOL)(6-14)
10Travis J. Hansen and Daniel A. Crowl. “Estimation of the Flammable Zone Boundaries for Flammable Gases.” Process Safety Progress 29 (June 2010): 3.
where
UOL is the upper oxygen limit (volume percent fuel in oxygen),
UFL is the upper flammable limit (volume percent fuel in air),
UFL0 is the oxygen concentration at the upper flammable limit (volume percent oxygen in air), and
CUOL is a fitting constant.
This equation requires only UFL data. Hansen and Crowl found a good fit to experimental data with Equation 6-14 for a number of fuels using CUOL = −1.87.
Example 6-5
Estimate the UOL for methane using Equation 6-14.
Solution
From Appendix B, the UFL for methane is 15.0 volume percent fuel in air, so UFL = 15%. If we select a basis of 100 moles of gas mixture, then 15 moles is methane and the remaining 85 moles is air. Of the 85 moles of air, (0.21)(85) = 17.85 moles of oxygen. Thus, UFLO = 17.85%. Substituting into Equation 6-14:
UOL=UFL[100−CUOL(100−UFL0)]UFL0+UFL(1−CUOL)=(15%)[100+1.87(100−17.85)]17.85%+(15%)(1+1.87)=62.4%
This compares to the experimental value of 61% shown in Table 6-2.
Limiting Oxygen Concentration (LOC) and Inerting
The LFL is based on fuel in air. However, oxygen is the key oxidizing ingredient and there is a minimum oxygen concentration required to propagate a flame. This is an especially useful result, because explosions and fires can be prevented by reducing the oxygen concentration regardless of the concentration of the fuel. This concept is the basis for a common procedure called inerting (see Chapter 7).
Below the limiting oxygen concentration (LOC), the reaction cannot generate enough energy to heat the entire mixture of gases (including the inert gases) to the extent required for the self-propagation of the flame. The LOC has also been called the minimum oxygen concentration (MOC) and the maximum safe oxygen concentration (MSOC), among other names.
Table 6-3 contains LOC values for a number of materials. The LOC depends on the inert gas species. It has units of percentage of moles of oxygen in total moles. If experimental data are not available, the LOC is estimated using the stoichiometry of the combustion reaction and the LFL. This procedure works for many hydrocarbons.
Table 6-3 Limiting Oxygen Concentrations (LOCs)
Gas or vapor |
N2/Air |
CO2/Air |
Gas or vapor |
N2/Air |
CO2/Air |
|---|---|---|---|---|---|
Methane |
12.1 |
14.6 |
Kerosene |
10 (150°C) |
13 (150°C) |
Ethane |
11 |
13.4 |
JP-1 fuel |
10.5 (150°C) |
14 (150°C) |
Propane |
11.4 |
14.3 |
JP-3 fuel |
12 |
14.5 |
n-Butane |
12.1 |
14.8 |
JP-4 fuel |
11.5 |
14.5 |
Isobutane |
12 |
14.8 |
Natural gas |
12 |
14.4 |
n-Pentane |
12.1 |
14.4 |
n-Butyl chloride |
14 |
– |
Isopentane |
12 |
14.5 |
|
12 (100°C) |
– |
n-Hexane |
11.9 |
14.5 |
Methylene chloride |
19 (30°C) |
– |
n-Heptane |
11.5 |
14.5 |
|
17 (100°C) |
– |
Ethylene |
10 |
11.7 |
Ethylene dichloride |
13 |
– |
Propylene |
11.5 |
14 |
|
11.5 (100°C) |
– |
1-Butene |
11.6 |
14 |
Methyl chloroform |
14 |
– |
Isobutylene |
12 |
15 |
Trichloroethylene |
9 (100°C) |
– |
Butadiene |
10.4 |
13.1 |
Acetone |
11.5 |
14 |
3-Methyl-l-butene |
11.5 |
14 |
i-butanol |
NA |
16.5 |
Benzene |
11.4 |
13.9 |
Carbon disulfide |
5 |
7.5 |
Toluene |
9.5 |
– |
Carbon monoxide |
5.5 |
5.5 |
Styrene |
9.0 |
– |
Ethanol |
10.5 |
13 |
Ethylbenzene |
9.0 |
– |
2-Ethyl butanol |
9.3 (150°C) |
– |
Vinyltoluene |
9.0 |
– |
Ethyl ether |
10.5 |
13 |
Diethylbenzene |
8.5 |
– |
Hydrogen |
5 |
5.2 |
Cyclopropane |
11.7 |
13.9 |
Hydrogen sulfide |
7.5 |
11.5 |
Gasoline |
|
|
Methanol |
10 |
12 |
(70/100) |
12 |
15 |
Methyl acetate |
11 |
13.5 |
(100/130) |
12 |
15 |
|
|
|
(115/145) |
12 |
14.5 |
|
|
|
Note: LOC is the volume percent oxygen concentration above which combustion can occur.
Source: Data from National Fire Protection Association. NFPA 69, Standard on Explosion Prevention (Quincy, MA: National Fire Protection Association, 2014).
Example 6-6
Estimate the LOC for butane (C4H10).
Solution
The stoichiometry for this reaction is
C4H10+6.5O2→4CO2+5H2O
The LFL for butane (from Appendix B) is 1.8% by volume. From the stoichiometry,
LOC=(moles fueltotal moles)(moles O2moles fuel)=LFL(moles O2moles fuel)
By substitution, we obtain
LOC= (1.8moles fueltotal moles)(6.5 moles O21.0 moles fuel)=11.7 vol. % O2
The combustion of butane is preventable by adding nitrogen, carbon dioxide, or even water vapor until the oxygen concentration is less than 11.7%. The addition of water, however, is not recommended because any condition that condenses water would move the oxygen concentration back into the flammable region.
Example 6-6 shows that the LOC can be estimated using the equation
LOC=z(LFL)(6-15)
Equation 6-15 does not produce very good results.
Hansen and Crowl11 found that a better estimate of the LOC is given by
LOC=(LFL−CLOCUFL1−CLOC)(UFL0UFL)(6-16)
11Travis J. Hansen and Daniel A. Crowl. “Estimation of the Flammable Zone Boundaries.” Process Safety Progress 29 (June 2010): 3.
where
LOC is the limiting oxygen concentration (percent oxygen),
LFL is the lower flammable limit (percent fuel in air),
UFL is the upper flammable limit (percent fuel in air),
UFL0 is the oxygen concentration at the upper flammable limit (volume percent oxygen in air), and
CLOC is a fitting constant.
Data analysis of numerous experimental values found that CLOC = –1.11 gave a good fit for many hydrocarbons.
Example 6-7
Estimate the LOC for butane using Equation 6-16. Compare this estimate to the results of Example 6-6.
Solution
From Appendix B, for butane, LFL = 1.8% and UFL = 8.5%. The oxygen concentration at the upper flammable limit is
UFL0=(0.21)(100−8.5)=19.21% oxygen
Substituting into Equation 6-16,
LOC=(LFL-CLOCUFL1-CLOC)(UFL0UFL)=[1.8 %+(1.11)(8.5%)1+1.11](19.21%8.5%)=12.0%
This compares to the experimental value of 12% shown in Table 6-3. Equation 6-15 produces a value of 11.7%, which is much lower than the experimental value.
Flammability Diagram
A general way to represent the flammability of a gas or vapor is by the triangle diagram shown in Figure 6-5. Concentrations of fuel, oxygen, and inert material (in volume or mole percent) are plotted on the three axes. Each apex of the triangle represents either 100% fuel, oxygen, or nitrogen. The tick marks on the scales show the direction in which the scale moves across the figure. Thus, point A represents a mixture composed of 60% methane, 20% oxygen, and 20% nitrogen. The zone enclosed by the dashed line represents all mixtures that are flammable. Because point A lies outside the flammable zone, a mixture of this composition is not flammable.

No. 3, 1998, pp 176–183.)
The air line represents all possible combinations of fuel plus air. It extends from the point where fuel is 0%, oxygen is 21%, and nitrogen is 79% to the point where fuel is 100%, oxygen is 0%, and nitrogen is 0%. The equation for this line is
Fuel% =−(10079)× nitrogen % +100(6-17)
The stoichiometric line represents all stoichiometric combinations of fuel plus oxygen. The combustion reaction can be written in the form
Fuel+zO2→combustion products(6-18)
where z is the stoichiometric coefficient for oxygen. The intersection of the stoichiometric line with the oxygen axis (in volume percent oxygen) is given by
100(z1+z)(6-19)
Equation 6-19 is derived by realizing that on the oxygen axis, no nitrogen is present. Thus the moles present equals fuel (1 mole) plus oxygen (z moles). The total moles is thus 1 + z, and the mole or volume percent of oxygen is given by Equation 6-15.
The stoichiometric line extends from a point where the fuel is 100/(1 + z), oxygen is 100z/(1 + z), and nitrogen is 0%, to a point where fuel is 0%, oxygen is 0%, and nitrogen is 100%. The equation for the stoichiometric line is
Fuel %=100−Nitrogen %(1+z)(6-20)
The LOC is also shown in Figure 6-5. Clearly, any gas mixture containing oxygen below the LOC is not flammable since the flammability zone does not extend below the LOC.
The shape and size of the flammability zone on a flammability diagram changes with a number of parameters, including fuel type, temperature, pressure, and inert species. Thus, the flammability limits and the LOC also change with these parameters.
Several rules and equations can be developed for working with flammability diagrams. More details on the development of these rules can be found on the website for this textbook. These rules are as follows:
If two gas mixtures R and S are combined, the resulting mixture composition lies on a line connecting the points R and S on the flammability diagram. The location of the final mixture on the straight line depends on the relative moles in the mixtures combined: If mixture S has more moles, the final mixture point will lie closer to point S. This is identical to the lever rule used for phase diagrams.
Figure 6-6 shows this rule and the following equation results:
xAM−xARxAS−xAM=xCM−xCRxCS−xCM(6-21)
xAM−xARxAS−xAM=xCM−xCRxCS−xCM(6-21) 
Figure 6-6 Flammability diagram for Rule 1 using Equation 6-21. If two mixtures are combined, the resulting mixture lies along a line between the two mixtures. If a mixture R is continuously diluted with mixture S, the mixture composition follows along the straight line between points R and S on the flammability diagram. As the dilution continues, the mixture composition moves closer and closer to point S. Eventually, at infinite dilution the mixture composition is at point S.
For systems having composition points that fall on a straight line passing through an apex corresponding to one pure component, the other two components are present in a fixed ratio along the entire line length.
Figure 6-7 shows this rule and the following equation results:
xAxB=x100−x(6-22)
xAxB=x100−x(6-22) 
Figure 6-7 Geometry for Rule 3 using Equation 6-2. The ratio of the components A and B is constant along the line shown and is given by x/(100 – x). The LOC can be estimated by reading the oxygen concentration at the intersection of the stoichiometric line and a horizontal line drawn through the LFL. This is equivalent to the equation
LOC=z(LFL)(6−15)
The following equations are used to transform triangle diagram coordinates (tA, tc) to rectangular coordinates (x, y):
y=tCsin(60π180)x=1−tA−ycot(60π180)(6-23)
where the sin and cot values in the parentheses are in degrees. These transformations are useful for drawing a triangle diagram using a spreadsheet.
Another interesting feature with triangle diagrams is that the three triangle legs can be any length and the rules still apply. In this case, Equation 6-23 must be modified accordingly.
These rules are useful for tracking the gas composition during a process operation to determine whether a flammable mixture exists during the procedure. For example, consider a storage vessel containing pure methane whose inside walls must be inspected as part of its periodic maintenance procedure. For this operation, the methane must be removed from the vessel and replaced by air for the inspection workers to breathe. The first step in the procedure is to depressurize the vessel to atmospheric pressure. At this point the vessel contains 100% methane, represented by point A in Figure 6-8. If the vessel is opened and air is allowed to enter and mix with the fuel, the composition of gas within the vessel will follow the air line in Figure 6-8 until the vessel gas composition eventually reaches point B, pure air. During this operation, the gas composition passes through the flammability zone. If an ignition source of sufficient strength were present, then a fire or explosion would result.

The procedure is reversed for placing the vessel back into service. In this case, the procedure begins at point B in Figure 6-8, with the vessel containing air. If the vessel is closed and methane is introduced and mixes with the air, then the gas composition inside the vessel will follow the air line and finish at point A. Again, the mixture is flammable as the gas composition moves through the flammability zone.
An inerting procedure can be used to avoid the flammability zone for both cases. This is discussed in more detail in Chapter 7.
The determination of a complete flammability diagram requires several hundred tests using a specific testing apparatus (see Figure 6-15 later in this chapter). Diagrams with experimental data for methane, ethylene, and hydrogen are shown in Figures 6-9, 6-10, and 6-11, respectively. Data in the center region of the flammability zone are not available because the maximum pressure exceeds the pressure rating of the test vessel. For these data, a mixture is considered flammable if the pressure increase after ignition is greater than 7% of the original ambient pressure, in accordance with ASTM E918.12 Note that many more data points are shown than are required to define the flammability limits. This was done to obtain a more complete understanding of the pressure versus time behavior of the combustion over a wide range of mixtures. This information is important for mitigation of an explosion.
12ASTM E918-83, Standard Practice for Determining Limits of Flammability of Chemicals at Elevated Temperature and Pressure (W. Conshocken, PA: ASTM, 2011).

dissertation, Michigan Technological University, 1999.)

dissertation, Michigan Technological University, 1999.)

Figure 6-11 is a different geometry from Figures 6-9 and 6-10 but still conveys the same information. Note that the hydrogen axis is diagonal, while the nitrogen and oxygen axes are rectangular. The LFL (about 4% fuel) is still shown as the lower intersection of the flammability zone with the air line, and the UFL (about 75% fuel) is the upper intersection of the flammability zone with the air line. The LOC is the oxygen line that just touches the flammability zone—in this case about 5% oxygen. Some people prefer this form of the triangle diagram since it is easier to plot—the nitrogen and oxygen are the x and y axes, respectively.
A number of important features are shown in Figures 6-9 to 6-11. First, the size of the flammability zone increases from methane to ethylene to hydrogen—the UFL is correspondingly higher. Second, the combustion of the methane and ethylene produces copious amounts of soot in the upper fuel-rich parts of the flammability zone. There is no soot with hydrogen because there is no carbon. Finally, the lower boundary of the flammability zone is mostly horizontal, and the LOL can be approximated by the LFL.
For most flammable materials, detailed experimental data of the type shown in Figures 6-9 to 6-11 are unavailable. Several methods have been developed to approximate the flammability zone:
Method 1 (Figure 6-12): Given the flammability limits in air, the LOC, and flammability limits in pure oxygen, the procedure is as follows:
Draw flammability limits in air as points on the air line.
Draw flammability limits in pure oxygen as points on the oxygen scale.
Use Equation 6-19 to locate the stoichiometric point on the oxygen axis, and draw the stoichiometric line from this point to the 100% nitrogen apex.
Locate the LOC on the oxygen axis, and draw a line parallel to the fuel axis until it intersects with the stoichiometric line. Draw a point at this intersection.
Connect all the points shown.

The flammability zone derived from this approach is only an approximation of the actual zone. Note that the lines defining the zone limits in Figures 6-9 to 6-11 are not exactly straight. This method also requires flammability limits in pure oxygen—data that are not readily available. Flammability limits in pure oxygen for a number of common hydrocarbons are provided in Table 6-2.
Method 2 (Figure 6-13): Given the flammability limits in air and the LOC, the procedure is as follows: Use steps 1, 3, and 4 from method 1. In this case, only the points at the nose of the flammability zone can be connected. The flammability zone from the air line to the oxygen axis cannot be detailed without additional data, although it extends all the way to the oxygen axis and typically expands in size. The lower boundary can also be approximated by the LFL.

Method 3 (Figure 6-14): Given the flammability limits in air, the procedure is as follows: Use steps 1 and 3 from method 1. Estimate the LOC using Equation 6-15 or 6-16. This is only an estimate, and usually (but not always) provides a conservative LOC.

Autoignition
The autoignition temperature (AIT) of a vapor, sometimes called the spontaneous ignition temperature (SIT), is the temperature at which the vapor ignites spontaneously. The autoignition temperature is a function of the concentration of vapor, volume of vapor, pressure of the system, presence of catalytic material, and flow conditions. It is essential to experimentally determine AITs at conditions as close as possible to process conditions.
Composition affects the AIT; rich or lean mixtures have higher AITs. Larger system volumes, increases in pressure, and increases in oxygen concentration all decrease AITs. This strong dependence on conditions illustrates the importance of exercising caution when using AIT data.
AIT data are provided in Appendix B.
Auto-oxidation
Auto-oxidation is the process of slow oxidation with accompanying evolution of heat, sometimes leading to autoignition if the energy is not removed from the system. Liquids with relatively low volatility are particularly susceptible to this problem. Liquids with high volatility are less susceptible to autoignition because they self-cool as a result of evaporation.
Many fires are initiated as a result of auto-oxidation, which is also referred to as spontaneous combustion. Some examples of auto-oxidation with a potential for spontaneous combustion include oil on a rag in a warm storage area, insulation on a steam pipe saturated with certain polymers, and filter aid saturated with certain polymers. In fact, cases have been recorded where 10-year-old filter aid residues were ignited when the land-filled material was bulldozed and exposed to air, allowing auto-oxidation and eventual autoignition. These examples illustrate why special precautions must be taken to prevent fires that can result from auto-oxidation and autoignition.
Adiabatic Compression
An additional means of ignition is adiabatic compression. For example, gasoline and air in an automobile cylinder will ignite if the vapors are compressed to an adiabatic temperature that exceeds the autoignition temperature. This is the cause of preignition knock in engines that are running too hot and too lean.
Several large accidents have been caused by flammable vapors being sucked into the intake of air compressors, with their subsequent compression resulting in autoignition. A compressor is particularly susceptible to autoignition if it has a fouled after-cooler. Safeguards must be included in the process design to prevent undesirable fires that can result from adiabatic compression.
The adiabatic temperature increase for an ideal gas is computed from the thermodynamic adiabatic compression equation:
Tf=Ti(PfPi)(γ−1)/γ(6-24)
where
Tf is the final absolute temperature,
Ti is the initial absolute temperature,
Pf is the final absolute pressure,
Pi is the initial absolute pressure, and
γ is the heat capacity ratio given by Cp / Cv.
The potential consequences of adiabatic temperature increases within a chemical plant are illustrated in the following two examples.
Example 6-8
What is the final temperature after compressing air over liquid hexane from 14.7 psia to 500 psia if the initial temperature is 100°F? The AIT of hexane is 487°C (Appendix B), and γ for air is 1.4.
Solution
From Equation 6-24, we have
Tf=(37.8+273)(50014.7)(0.4/1.4)=851 K=578°C
This temperature exceeds the AIT for hexane, so ignition should be expected.
These examples illustrate the importance of careful design, careful monitoring of conditions, and periodic preventive maintenance programs when working with flammable gases and compressors. This is especially important today, because high-pressure process conditions are becoming more common in modern chemical plants.
Example 6-9
The lubricating oil in piston-type compressors is always found in minute amounts on the cylinder bore. Compressor operations must always be maintained well below the AIT of the oil to prevent explosion.
A particular lubricating oil has an AIT of 400°C. Compute the compression ratio required to raise the temperature of air to the AIT of this oil. Assume an initial air temperature of 25°C and 1 atm.
Solution
Equation 6-24 applies. Solving for the compression ratio, we obtain
(PfPi)=(TfTi)γ(γ−1)=(400+27325+273)1.4/0.4=17.3
This ratio represents an output pressure of only (17.3)(14.7 psia) = 254 psia. The actual compression ratio or pressure should be kept well below this level.
6-5 Flammability Characteristics of Dusts13
13Rolf K. Eckhoff. Dust Explosions in the Process Industries: Identification, Assessment and Control of Dust Hazards, 3rd ed. (Amsterdam, Netherlands: Gulf Professional Publishing) 2003.
Dusts can present a significant flammability and explosion hazard. Dust explosions occur when finely divided particles of solid material are dispersed in air and ignited. The dust particles can be either an unwanted by-product or the product itself. Dusts are typically defined as a solid mixture with a maximum particle size of less than 500 microns (µm).
Many common materials, such as metals, wood, and agricultural products, become highly flammable and even explosive when in dust form. Explosions involving dusts are most common in the flour milling, grain storage, and coal mining industries. Accidents involving dust explosions can be quite substantial: The Imperial Sugar explosion in Wentworth, Georgia, resulted in 14 fatalities.14
14“Imperial Sugar Dust Explosion and Fire: US Chemical Safety Board Report,” September 24, 2009. csb.gov.
An initial dust explosion can cause secondary explosions. The primary explosion sends a pressure or shock wave through the plant, stirring up additional dust, and possibly resulting in a secondary explosion. In this fashion, the explosion leapfrogs its way through a plant. Many times the secondary explosions are more damaging than the primary explosion.
Dust explosions are even more difficult to characterize than gaseous explosions because more parameters are associated with dust explosions. For dusts, deflagrations appear to be much more common than detonations.15 The pressure waves from dust deflagrations, however, are powerful enough to destroy structures and kill or injure people.
15Frank P. Lees. Loss Prevention in the Process Industries, 2nd ed. (Boston, MA: Butterworths, 1996), p. 17/265.
To be explosive, a dust mixture must have the following characteristics:
The particles must be smaller than a certain maximum size, typically less than 400 microns.
The dust loading must be reasonably uniform via adequate dispersion.
The dust must have a certain amount of confinement.
The particle loading must be between certain limits.
Frequently the three legs of the fire triangle—fuel, oxygen, and ignition source—are combined with confinement and dispersion to form a dust pentagon.
For many dusts,16 the lower explosion limit is between 20 g/m3 and 60 g/m3 and the upper explosion limit is between 2 kg/m3 and 6 kg/m3.
16W. Bartknecht. Explosions: Course, Prevention, Protection, (Berlin; Springer-Verlag, 1981), p. 27.
6-6 Sprays and Mists17
17Frank P. Lees. Loss Prevention in the Process Industries, 2nd ed. (Boston, MA: Butterworths, 1996).
Mists and sprays also affect flammability limits.18 For suspensions with drop diameters less than 0.01 mm, the LFL is virtually the same as the substance in vapor form. This is true even at low temperatures where the liquid is nonvolatile and no vapor is present. Mists of this type can be formed by condensation.
18J. H. Borgoyne. “The Flammability of Mists and Sprays.” Chemical Process Hazards 2 (1965): 1.
For mechanically formed mists with drop diameters between 0.01 mm and 0.2 mm, the LFL decreases as the drop diameter increases. In experiments with larger drop diameters, the LFL was less than one-tenth of the normal LFL. This is important when inerting in the presence of mists.
When sprays have drop diameters between 0.6 mm and 1.5 mm, flame propagation is not likely. In this situation, however, the presence of small drops and disturbances that shatter the larger drops may create a hazardous condition.
6-7 Ignition Energy
The minimum ignition energy (MIE) is the minimum energy input required to initiate combustion. All flammable materials (including dusts) have MIEs. The MIE depends on the specific chemical or mixture, the concentration, pressure, and temperature. A few MIEs are given in Table 6-4. Most MIEs are reported for fuel in air.
Table 6-4 Minimum Ignition Energy for Selected Gases
Chemical |
Minimum ignition energy (mJ) |
|---|---|
Acetylene |
0.020 |
Benzene |
0.225 |
1,3-Butadiene |
0.125 |
n-Butane |
0.260 |
Cyclohexane |
0.223 |
Cyclopropane |
0.180 |
Ethane |
0.240 |
Ethene |
0.124 |
Ethylacetate |
0.480 |
Ethylene oxide |
0.062 |
n-Heptane |
0.240 |
Hexane |
0.248 |
Hydrogen |
0.018 |
Methane |
0.280 |
Methanol |
0.140 |
Methyl acetylene |
0.120 |
Methyl ethyl ketone |
0.280 |
n-Pentane |
0.220 |
2-Pentane |
0.180 |
Propane |
0.250 |
Source: Data from I. Glassman. Combustion, 3rd ed. (New York: Academic Press, 1996).
Experimental data for flammable vapors shows that,
As the pressure increases, the MIE decreases.
As temperature increases, the MIE decreases.
Starting from low fuel concentration, as the fuel concentration increases, the ignition energy decreases to a minimum (the MIE) and then increases.
An increase in the nitrogen concentration increases the MIE.
For flammable dusts, the MIE is, in general, at energy levels somewhat higher than combustible gases.
Many hydrocarbons have MIEs of about 0.25 mJ. This is low compared with sources of ignition. For example, a static discharge of 22 mJ is initiated by walking across a rug, and an ordinary spark plug has a discharge energy of 25 mJ or higher. Electrostatic discharges, as a result of fluid flow, also have energy levels exceeding the MIEs of flammable materials and can provide an ignition source, contributing to explosions (see Chapter 7).
6-8 Ignition Sources19
19Accident Prevention Manual for Industrial Operations (Chicago, IL: National Safety Council, 1974).
Fires and explosions can be prevented by eliminating ignition sources, as demonstrated by removing one leg of the fire triangle shown in Figure 6-1. Various ignition sources were tabulated for more than 25,000 fires by the Factory Mutual Engineering Corporation and are summarized in Table 6-5. The sources of ignition are numerous; consequently, it is impossible to identify and eliminate them all. The main reason for rendering a flammable liquid inert is to prevent a fire or explosion by ignition from an unidentified source. Because all sources of ignition are not likely to be identified, engineers must still continue to identify and eliminate them.
Table 6-5 Ignition Sources of Major Fires
Electrical (wiring of motors) |
23% |
Smoking |
18% |
Friction (bearings or broken parts) |
10% |
Overheated materials (abnormally high temperatures) |
8% |
Hot surfaces (heat from boilers, lamps, etc.) |
7% |
Burner flames (improper use of torches, etc.) |
7% |
Combustion sparks (sparks and embers) |
5% |
Spontaneous ignition (rubbish, etc.) |
4% |
Cutting and welding (sparks, arcs, heat, etc.) |
4% |
Exposure (fires jumping into new areas) |
3% |
Incendiarism (fires maliciously set) |
3% |
Mechanical sparks (grinders, crushers, etc.) |
2% |
Molten substances (hot spills) |
2% |
Chemical action (processes not in control) |
1% |
Static sparks (release of accumulated energy) |
1% |
Lightning (where lightning rods are not used) |
1% |
Miscellaneous |
1% |
Source: Accident Prevention Manual for Industrial Operations (Chicago, IL: National Safety Council, 1974).
Some special situations might occur in a process facility where it is impossible to avoid flammable mixtures. In these cases, a detailed and thorough safety analysis is required to eliminate as many ignition sources as possible to achieve an acceptable level of safety.
The elimination of the ignition sources with the greatest probability of occurrence (see Table 6-5) should be given the highest attention. Combinations of sources must also be investigated. The goal is to eliminate or minimize ignition sources because the probability of a fire or explosion increases rapidly as the number of ignition sources increases. The effort required increases significantly as the size of the plant increases; potential ignition sources may be in the thousands.
6-9 Experimental Characterization of Gas/Vapor and Dust Explosions
Gases/Vapors
The apparatus used to characterize the explosive behavior of gases and vapors is shown in Figure 6-15. This apparatus consists of a spherical pressure vessel with an adequate pressure rating to withstand 252the maximum pressure of the explosion. A vessel volume of 10 to 20 liters is typically used. A high-precision pressure gauge is used to measure the pressure during the gas mixing as well as during the explosion. A temperature gauge measures the temperature since the results are dependent on the temperature. The mixing bar is used to mix the gases. A fuse wire is typically used to ignite the gas, consisting of a 1-cm 40-gauge wire. In most cases, a computer is used to operate the experiment and collect the data.

The experimental procedure is as follows. The apparatus is initially evacuated. Gases are then metered into the vessel through the gas manifold, with the concentrations being controlled by measuring the partial pressure from the pressure gauge. The mixing bar is active during the gas addition. The mixer is then turned off. After a short time delay to let the gas reach equilibrium, the igniter is triggered and the explosion is initiated. The results are highly dependent on gas composition, so it is important to use ultra-high-purity gas sources.
Since a deflagration occurs, the pressure in the vessel is uniform across the volume during the explosion. Measurement of the combustion pressure on the sphere surface provides information on the progress of the explosion.
Two parameters are used to characterize the behavior of gas/vapor explosions: the maximum pressure and the maximum pressure rate. The maximum pressure is indicative of the combustion equilibrium, while the maximum pressure rate is indicative of the flame front’s propagation speed and is representative of the robustness of the explosion. Clearly, higher values for these parameters indicate an increased explosion hazard.
Figure 6-16 shows how these two parameters are determined experimentally from pressure–time data for a single experiment run at a specific concentration. After ignition of the gas/vapor at t = 0, the pressure increases rapidly to a peak value; it then decreases at a much slower rate due to quenching and cooling from the vessel surface. The maximum pressure is easily determined from the peak pressure. The maximum pressure rate is determined from the maximum slope of the pressure time curve. In this case, the maximum pressure is 8.5 bar and the maximum pressure rate is 316 bar/s.

The experiment is repeated at different concentrations, with the maximum pressure and maximum pressure rates being determined at each concentration. The highest maximum pressure and pressure rates do not necessarily occur at the same concentration.
The flammable limits for this fuel are determined from the maximum pressure versus concentration plot shown in Figure 6-17. Usually a criterion of a 7% pressure increase is used to define these limits. For the data shown, the lower flammable limit is 4.9% and the upper limit is 16.2%.

A plot of the logarithm of the maximum pressure slope versus the logarithm of the vessel volume frequently produces a straight line of slope −1/3, as shown in Figure 6-18. This relationship is called the cubic law and is represented by the following equations:

For gases: (dPdt)maxV1/3=constant =KG(6-25)
For dusts: (dPdt)maxV1/3=KSt(6-26)
where KG and KSt are the deflagration indexes for gas and dust, respectively. The subscript “St” for the dust deflagration index comes from the German word Staub, meaning “dust.”
Figure 6-19 shows how the deflagration index, KG, varies with composition for methane.
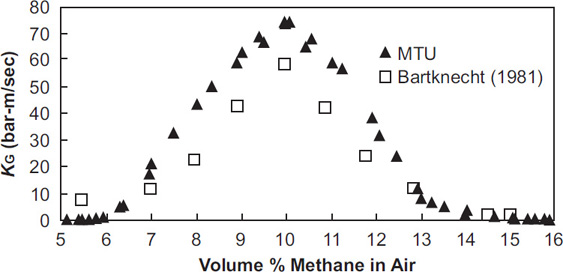
As the robustness of an explosion increases, the deflagration indexes KG and KSt increase. The cubic law states that the pressure front takes longer to propagate through a larger vessel. Pmax and KG data for vapors are shown in Table 6-6. Table 6-6 shows that good agreement is found between different investigators for the maximum pressure but that only limited agreement is found for the KG values. KG values are sensitive to experimental configuration and conditions.
Table 6-6 Maximum Pressures and Deflagration Indexes for a Number of Gases and Vapors
|
Maximum pressure Pmax (bar g) |
Deflagration index KG (bar-m/s) |
||||
Chemical |
NFPA 68 (2018) |
Bartknecht (1993) |
Senecal and Beaulieu (1998) |
NFPA 68 (1997) |
Bartknecht (1993) |
Senecal and Beaulieu (1998) |
|---|---|---|---|---|---|---|
Acetylene |
10.6 |
– |
– |
109 |
– |
– |
Ammonia |
5.4 |
– |
– |
10 |
– |
– |
Butane |
8.0 |
8.0 |
– |
92 |
92 |
– |
Carbon disulfide |
6.4 |
– |
– |
105 |
– |
– |
Diethyl ether |
8.1 |
– |
– |
115 |
– |
– |
Ethane |
7.8 |
7.8 |
7.4 |
106 |
106 |
78 |
Ethyl alcohol |
7.0 |
– |
– |
78 |
– |
– |
Ethyl benzene |
7.4 |
7.4 |
– |
94 |
96 |
– |
Ethylene |
– |
– |
8.0 |
– |
– |
171 |
Hydrogen |
6.8 |
6.8 |
6.5 |
659 |
550 |
638 |
Hydrogen sulfide |
7.4 |
– |
|
45 |
– |
– |
Isobutane |
|
|
7.4 |
– |
– |
67 |
Methane |
7.1 |
7.1 |
6.7 |
64 |
55 |
46 |
Methyl alcohol |
7.5 |
7.5 |
7.2 |
– |
75 |
94 |
Methylene chloride |
5.0 |
– |
– |
5 |
– |
– |
Pentane |
7.8 |
7.8 |
– |
104 |
104 |
– |
Propane |
7.9 |
7.9 |
7.2 |
96 |
100 |
76 |
Toluene |
7.8 |
7.8 |
– |
– |
94 |
– |
Data sources:
NFPA 68, Venting of Deflagrations (Quincy, MA: National Fire Protection Association, 1997).
NFPA 68, Standard on Explosion Protection (Quincy, MA: National Fire Protection Association, 2018).
W. Bartknecht. Explosions-Schutz: Grundlagen und Anwendung (New York, NY: Springer-Verlag, 1993).
J. A. Senecal and P. A. Beaulieu. “KG: Data and Analysis.” In 31st Loss Prevention Symposium (New York, NY: American Institute of Chemical Engineers, 1997).
Dusts
The experimental apparatus used to characterize the explosive nature of dusts is shown in Figure 6-20. This device is similar to the vapor explosion apparatus, with the exception of a larger volume—typically 20 L or larger—and the addition of a sample container and a dust distribution ring. The distribution ring ensures proper distribution and mixing of the dust before ignition.

The experimental procedure is as follows. The dust sample is placed in the sample container. The computer system opens the solenoid valve, and the dust is driven by air pressure from the sample container through the distribution ring and into the dust sphere. After a delay of several milliseconds to ensure proper mixing and distribution of the dust, the ignitor is discharged. The computer measures the pressure as a function of time using high- and low-speed pressure transducers. The air used to drive the dust into the sphere is carefully metered to ensure a pressure of 1 atm (1.013 bar) within the sphere at ignition time. A typical pressure versus time plot from the dust explosion apparatus is shown in Figure 6-21.

The data are collected and analyzed in the same fashion as for the vapor explosion apparatus. The maximum pressure and the maximum rate of pressure increase are determined, as well as the flammability limits.
Table 6-7 shows experimental data for dusts. This includes the maximum pressure during combustion, Pmax and the deflagration index, KSt, using Equation 6-26. Notice at the top of Table 6-7 that dusts are also classified into four St-classes depending on the value of the deflagration index. The higher the deflagration index, the higher the St-class.
Table 6-7 St-Classes for Dusts and Combustion Data for Dust Clouds
|
Deflagration index, KSt (bar m/s) |
St-class |
|
|
|
|---|---|---|---|---|---|
|
0 |
St-0 |
|
|
|
|
1–200 |
St-1 |
|
|
|
|
200–300 |
St-2 |
|
|
|
|
>300 |
St-3 |
|
|
|
Dust |
Median particle size (μm) |
Minimum explosive dust concentration (g/m3) |
Pmax (bar g) |
KSt (bar-m/s) |
Minimum ignition energy (mJ) |
Cotton, wood, peat |
|
|
|
|
|
Cotton |
44 |
100 |
7.2 |
24 |
– |
Cellulose |
51 |
60 |
9.3 |
66 |
250 |
Wood dust |
33 |
– |
– |
– |
100 |
Wood dust |
80 |
– |
– |
– |
7 |
Paper dust |
<10 |
– |
5.7 |
18 |
– |
Feed, food |
|
|
|
|
|
Dextrose |
80 |
60 |
4.3 |
18 |
– |
Fructose |
200 |
125 |
6.4 |
27 |
180 |
Fructose |
400 |
– |
– |
– |
>4000 |
Wheat grain dust |
80 |
60 |
9.3 |
112 |
– |
Milk powder |
165 |
60 |
8.1 |
90 |
75 |
Rice flour |
– |
60 |
7.4 |
57 |
>100 |
Wheat flour |
50 |
– |
– |
– |
540 |
Milk sugar |
10 |
60 |
8.3 |
75 |
14 |
Coal, coal products |
|
|
|
|
|
Activated carbon |
18 |
60 |
8.8 |
44 |
– |
Bituminous coal |
<10 |
– |
9.0 |
55 |
– |
Plastics, resins, rubber |
|
|
|
|
|
Polyacrylamide |
10 |
250 |
5.9 |
12 |
– |
Polyester |
<10 |
– |
10.1 |
194 |
– |
Polyethylene |
72 |
– |
7.5 |
67 |
– |
Polyethylene |
280 |
– |
6.2 |
20 |
– |
Polypropylene |
25 |
30 |
8.4 |
101 |
– |
Polypropylene |
162 |
200 |
7.7 |
38 |
– |
Polystyrene (copolymer) |
155 |
30 |
8.4 |
110 |
– |
Polystyrene (hard foam) |
760 |
– |
8.4 |
23 |
– |
Polyurethane |
3 |
<30 |
7.8 |
156 |
– |
Intermediate products, auxiliary materials |
|
|
|
|
|
Adipinic acid |
<10 |
60 |
8.0 |
97 |
– |
Naphthalene |
95 |
15 |
8.5 |
178 |
<1 |
Salicylic acid |
– |
30 |
– |
– |
– |
Other technical, chemical products |
|
|
|
|
|
Organic dyestuff (blue) |
<10 |
– |
9.0 |
73 |
– |
Organic dyestuff (red) |
<10 |
50 |
11.2 |
249 |
– |
Organic dyestuff (red) |
52 |
60 |
9.8 |
237 |
– |
Metals, alloys |
|
|
|
|
|
Aluminum powder |
<10 |
60 |
11.2 |
515 |
– |
Aluminum powder |
22 |
30 |
11.5 |
110 |
– |
Bronze powder |
18 |
750 |
4.1 |
31 |
– |
Iron (from dry filter) |
12 |
500 |
5.2 |
50 |
– |
Magnesium |
28 |
30 |
17.5 |
508 |
– |
Magnesium |
240 |
500 |
7.0 |
12 |
– |
Silicon |
<10 |
125 |
10.2 |
126 |
54 |
Zinc (dust from collector) |
<10 |
250 |
6.7 |
125 |
– |
Other inorganic products |
|
|
|
|
|
Graphite (99.5% C) |
7 |
<30 |
5.9 |
71 |
– |
Sulfur |
20 |
30 |
6.8 |
151 |
– |
Toner |
<10 |
60 |
8.9 |
196 |
4 |
Source: Data selected from R. K. Eckoff. Dust Explosions in the Process Industries (Oxford, UK: Butterworth-Heinemann, 1997). A 2003 edition is also available.
Application of Flammability Data of Gases/Vapors and Dusts
Characterization of the explosive behavior of gases, vapors, and dusts is highly dependent on the apparatus and procedure. The results are not fundamentally based, unlike the case for heat capacity or density. Thus, experimental flammability data should always be obtained at conditions as close as possible to the actual process conditions and those data should be applied using proper engineering judgment.
The explosion characteristics determined using the vapor and dust explosion apparatus are used to operate the process safely, as shown below. Note that these experimental parameters are not an absolute boundary between safe and unsafe operation so suitable safety margins must be applied.
The flammable limits are used to determine the safe concentrations for operation or the quantity of inert material required to control the concentration within safe regions.
The maximum rate of pressure increase indicates the robustness of an explosion. Thus, the explosive behavior of different materials can be compared on a relative basis. The maximum rate is also used to design a vent for relieving a vessel during an explosion before the pressure ruptures the vessel or to establish the time interval for adding an explosion suppressant (water, carbon dioxide, or other) to stop the combustion process.
6-10 Explosions
Explosion behavior is difficult to characterize, because it depends on a large number of parameters, including those shown in Table 6-8. Many approaches to the problem have been undertaken, including theoretical, semiempirical, and empirical methods. Despite these efforts, explosion behavior is still not completely understood. Practicing engineers, therefore, should use extrapolated results cautiously and provide a suitable margin of safety in all designs.
Table 6-8 Parameters Significantly Affecting the Behavior of Explosions
Ambient temperature |
Ambient pressure |
Composition of explosive material |
Physical properties of explosive material |
Nature of ignition source: type, energy, and duration |
Geometry of surroundings: confined or unconfined Congestion due to process equipment, piping, and other components |
Amount of combustible material |
Turbulence of combustible material |
Time before ignition |
Rate at which combustible material is released |
An explosion results from the rapid release of energy. This energy release must be sudden enough to cause a local accumulation of energy at the site of the explosion. The energy is then dissipated by a variety of mechanisms, including formation of a pressure wave, projectiles, thermal radiation, and acoustic energy. The damage from an explosion is caused by the dissipating energy.
If the explosion occurs in a gas, the sudden energy release causes the gas to expand rapidly, forcing back the surrounding gas and initiating a pressure wave that moves rapidly outward from the blast source. The pressure wave contains energy, which results in damage to the surroundings. In chemical plants, much of the damage from explosions is due to this pressure wave. Thus, to understand explosion impacts, we must understand the dynamics of the pressure wave.
A pressure wave propagating in air is called a blast wave because the pressure wave is followed by a strong wind. A shock wave or shock front results if the pressure front has an very abrupt pressure change. A shock wave is expected from highly explosive materials, such as TNT, but it can also occur from the sudden rupture of a pressure vessel. The maximum pressure in the shock or pressure wave over ambient pressure is called the peak overpressure.
Detonation and Deflagration
The damage from an explosion largely depends on whether the explosion results from a detonation or a deflagration. The difference between the two reflects whether the reaction front propagates above or below the speed of sound in the unreacted gases. For ideal gases, the speed of sound or sonic velocity is a function of temperature only and has a value of 344 m/s (1129 ft/s) at 20°C. Fundamentally, the sonic velocity is the speed at which information is transmitted through a gas.
In some combustion reactions, the reaction front is propagated by a strong pressure wave, which compresses the unreacted mixture in front of the reaction front above its autoignition temperature. This compression occurs rapidly, resulting in an abrupt pressure change or shock in front of the reaction front. Such an event is classified as a detonation, and it results in a reaction front and leading shock wave that propagates into the unreacted mixture at or above the sonic velocity of the unreacted gas.
For a deflagration, the energy from the reaction is transferred to the unreacted mixture by heat conduction and molecular diffusion. These processes are relatively slow, causing the reaction front to propagate at a speed less than the sonic velocity in the unreacted gas.
Figure 6-22 shows the physical differences between a detonation and a deflagration for a combustion reaction that occurs in the gas phase in the open. For a detonation, the reaction front moves at a speed greater than the speed of sound. A shock front is found a short distance in front of the reaction front. The reaction front provides the energy for the shock front and continues to drive it at sonic or higher speeds.

For a deflagration, the reaction front propagates at a speed less than the speed of sound. However, the pressure front moves at the speed of sound in the unreacted gas and moves away from the reaction front. One way to conceptualize the resulting pressure front is to consider the reaction front as producing a series of individual pressure fronts. These pressure fronts move away from the reaction front at the speed of sound and accumulate to form the main pressure front. The main pressure front will continue to grow in size as additional energy and pressure fronts are produced by the reaction front.
The pressure fronts produced by detonations and deflagrations are markedly different. A detonation produces a shock front, with an abrupt pressure rise, a maximum pressure of greater than 10 atm, and total duration that is typically less than 1 ms. The pressure front resulting from a deflagration is characteristically wide (many milliseconds in duration), flat (without an abrupt shock front), and with a maximum pressure much lower than the maximum pressure for a detonation (typically 6 to 10 times the initial pressure).
The behaviors of the reaction and pressure fronts differ from those shown in Figure 6-22 depending on the local geometry constraining the fronts. Different behavior occurs if the fronts propagate in a closed vessel, in a pipeline, or through a congested process unit. The gas dynamic behavior for complex geometries is beyond the scope of this text.
A deflagration can also evolve into a detonation—a phenomenon called a deflagration to detonation transition (DDT). This transition is particularly common in pipes but is unlikely to occur in vessels or open spaces. In a piping system, energy from a deflagration can feed forward to the pressure wave, resulting in an increase in the adiabatic pressure rise. The pressure then builds and eventually results in a full detonation.
Confined Explosions
A confined explosion occurs in a confined space, such as a vessel or a building. The two most common confined explosion scenarios involve explosive vapors and explosive dusts. Empirical studies have shown that the nature of the explosion is a function of several experimentally determined characteristics, which depend on the explosive material used. These characteristics include flammability or explosive limits, the rate of pressure rise after the flammable mixture is ignited, and the maximum pressure after ignition. They are determined using two similar laboratory devices, shown in Figures 6-15 and 6-20.
Blast Damage Resulting from Overpressure
The explosion of a dust or gas (either as a deflagration or a detonation) results in a reaction front moving outward from the ignition source preceded by a shock wave or pressure front. After the combustible material is consumed, the reaction front terminates, but the pressure wave continues its outward movement. A blast wave, which is composed of the pressure wave and subsequent wind, causes most of the damage in such an explosion.
Figure 6-23 shows the variation in pressure with time for a typical shock wave at a fixed location some distance from the explosion site. The explosion occurs at time t0. There exists a small but finite time t1 before the shock front travels from its explosive origin to the affected location. This time, t1, is called the arrival time. At t1, the shock front has arrived and a peak overpressure is observed, immediately followed by a strong transient wind. The pressure quickly decreases to ambient pressure at time t2, but the wind continues in the same direction for a short time. The time period t1 to t2 is called the shock duration. The shock duration is the period of greatest destruction to free-standing structures, so its value is important for estimating damage. The decreasing pressure continues to drop below ambient pressure to a maximum underpressure at time t3. For most of the underpressure period from t2 to t4, the blast wind reverses direction and flows toward the explosive origin. Although the underpressure period is associated with some damage, it is much less than the damage associated with the overpressure period because the maximum underpressure is only a few psi for typical explosions. The underpressure for large explosions and nuclear explosions, however, can be quite large, resulting in considerable damage. After attaining the maximum underpressure at time t3, the pressure will approach ambient pressure at time t4. At this time, the blast wind has moved beyond the reference point.

An important consideration is how the pressure is measured as the blast wave passes. If the pressure transducer is at right angles to the blast wave, the overpressure measured is called the side-on overpressure (sometimes called the free-field overpressure). At a fixed location, shown in Figure 6-23, the side-on overpressure increases abruptly to its maximum value (peak side-on overpressure) and then drops off as the blast wave passes. If the pressure transducer is placed facing toward the oncoming shock wave, then the measured pressure is the reflected overpressure. The reflected overpressure includes the side-on overpressure and the stagnation pressure. The stagnation pressure is caused by deceleration of the moving gas as it impacts the pressure transducer. The reflected pressure for low side-on overpressures is about twice the side-on overpressure and can reach as high as eight or more times the side-on overpressure for strong shocks. The reflected overpressure reaches its maximum when the blast wave arrives normal to the wall or object of concern, but decreases as the angle changes from normal. Many references report overpressure data without clearly stating how the overpressure is measured. In general, overpressure implies the side-on overpressure and frequently the peak side-on overpressure.
Blast damage is based on the determination of the peak side-on overpressure resulting from the pressure wave impacting a structure. In general, the damage is also a function of the rate of pressure increase and the duration of the blast wave. Good estimates of blast damage, however, are obtained using just the peak side-on overpressure.
Damage estimates based on overpressures are given in Table 6-9. As illustrated, significant damage is expected for even small overpressures.
Table 6-9 Damage Estimates for Common Structures Based on Overpressure
Overpressure |
|
|
|---|---|---|
psig |
kPa |
Damage |
0.02 |
0.14 |
Annoying noise (137 dB if of low frequency, 10–15 Hz) |
0.03 |
0.21 |
Occasional breaking of large glass windows already under strain |
0.04 |
0.28 |
Loud noise (143 dB), sonic boom, glass failure |
0.1 |
0.69 |
Breakage of small windows under strain |
0.15 |
1.03 |
Typical pressure for glass breakage |
0.3 |
2.07 |
“Safe distance” (probability 0.95 of no serious damage below this value); projectile limit; some damage to house ceilings; 10% window glass broken |
0.4 |
2.76 |
Limited minor structural damage |
0.5–1.0 |
3.4–6.9 |
Large and small windows usually shatter; occasional damage to window frames |
0.7 |
4.8 |
Minor damage to house structures |
1.0 |
6.9 |
Partial demolition of houses, made uninhabitable |
1–2 |
6.9–13.8 |
Corrugated asbestos shatters; corrugated steel or aluminum panels, fastenings fail, followed by buckling; wood panels (standard housing), fastenings fail, panels blow in |
1.3 |
9.0 |
Steel frame of clad building slightly distorted |
2 |
13.8 |
Partial collapse of walls and roofs of houses |
2–3 |
13.8–20.7 |
Concrete or cinder block walls, not reinforced, shatter |
2.3 |
15.8 |
Lower limit of serious structural damage |
2.5 |
17.2 |
50% destruction of brickwork of houses |
3 |
20.7 |
Heavy machines (3000 lb) in industrial buildings suffer little damage; steel frame buildings distort and pull away from foundations |
3–4 |
20.7–27.6 |
Frameless, self-framing steel panel buildings demolished; rupture of oil storage tanks |
4 |
27.6 |
Cladding of light industrial buildings ruptures |
5 |
34.5 |
Wooden utility poles snap; tall hydraulic presses (40,000 lb) in buildings slightly damaged |
5–7 |
34.5–48.2 |
Nearly complete destruction of houses |
7 |
48.2 |
Loaded train wagons overturned |
7–8 |
48.2–55.1 |
Brick panels, 8–12 in. thick, not reinforced, fail by shearing or flexure |
9 |
62.0 |
Loaded train boxcars completely demolished |
10 |
68.9 |
Probable total destruction of buildings; heavy machine tools (7000 lb) moved and badly damaged, very heavy machine tools (12,000 lb) survive |
300 |
2068 |
Limit of crater lip |
Note: These values are approximations.
Source: V. J. Clancey. “Diagnostic Features of Explosion Damage,” paper presented at the Sixth International Meeting of Forensic Sciences, Edinburgh, UK, 1972.
Experiments with explosives have demonstrated that the overpressure can be estimated using an equivalent mass of TNT, denoted as mTNT, and the distance from the ground-zero point of the explosion, denoted as r.20 The empirically derived scaling law is
20W. E. Baker. Explosions in Air (Austin: University of Texas Press, 1973); S. Glasstone. The Effects of Nuclear Weapons (Washington, DC: U.S. Atomic Energy Commission, 1962).
ze=rm1/3TNT(6-27)
The equivalent energy of TNT is 1120 cal/g.
Figure 6-24 provides a correlation for the scaled overpressure ps versus scaled distance ze with units of m/kg1/3. To convert ft/lb1/3 to m/kg1/3, multiply by 0.3967. The scaled overpressure ps is given by
ps = p0pa(6-28)

where
ps is the scaled overpressure (unitless),
p0 is the peak side-on overpressure (gauge), and
pa is the ambient pressure (absolute).
The data in Figure 6-24 are valid only for TNT explosions occurring on a flat surface. For explosions occurring in the open air, well above the ground, the resulting overpressures from Figure 6-24 are multiplied by 0.5. Most explosions occurring in chemical plants are considered to originate on the ground.
The data in Figure 6-24 are represented by the empirical equation
p0pa = 1616[1+(ze4.5)2]√1+(ze0.048)2√1+(ze0.32)2√1+(ze1.35)2(6-29)
The procedure for estimating the overpressure at any distance r resulting from the explosion of a mass of TNT is as follows: (1) use the scaling law and the correlations of Figure 6-24 to estimate the overpressure, and (2) use Table 6-9 to estimate the damage.
Example 6-10
One kilogram of TNT is exploded. Compute the overpressure at a distance of 30 m from the explosion.
Solution
The value of the scaling parameter is determined using Equation 6-27:
ze=rm1/3TNT=30 m(1.0 kg)1/3 = 30 m kg−1/3
From Figure 6-24, the scaled overpressure is 0.055. Thus, if the ambient pressure is 1 atm, then the resulting side-on overpressure is estimated at (0.055) (101.3 kPa) = 5.6 kPa (0.81 psi). Based on the data in Table 6-9, it appears that this overpressure will cause minor damage to house structures.
TNT Equivalency
TNT equivalency is a simple method for equating a known energy of a combustible fuel to an equivalent mass of TNT. The approach is based on the assumption that an exploding fuel mass behaves like exploding TNT on an equivalent energy basis. The equivalent mass of TNT is estimated using the following equation:
mTNT=ηmΔHcETNT(6-30)
where
mTNT is the equivalent mass of TNT (mass),
η is the empirical explosion efficiency (unitless),
m is the mass of hydrocarbon (mass),
ΔHc is the lower heat of combustion of the flammable gas (see Appendix B [energy/mass]), and
ETNT is the energy of explosion of TNT (energy/mass).
A typical value for the energy of explosion of TNT is 1120 cal/g = 4686 kJ/kg = 2016 Btu/lb
The explosion efficiency is one of the major problems in the equivalency method. The explosion efficiency is used to adjust the estimate for a number of factors, including incomplete mixing with air of the combustible material and incomplete conversion of the thermal energy to mechanical energy. The explosion efficiency is empirical, with most flammable cloud estimates varying between 1% and 10%, as reported by a number of sources. Explosion efficiencies can also be defined for solid materials, such as ammonium nitrate. A frequently used value in such cases is 2%.
The TNT equivalency method also uses an overpressure curve that applies to point-source detonations of TNT. Vapor cloud explosions (VCEs) are explosions that occur because of the release of flammable vapor over a large volume and are most commonly deflagrations. Because the TNT equivalency method is unable to consider the effects of flame speed acceleration resulting from confinement or congestion from process equipment, the overpressure curve for TNT tends to overpredict the overpressure near the VCE and to underpredict it at distances away from the VCE.
A key advantage of the TNT equivalency method is that it is easy to apply because the calculations are simple. The procedure to estimate the damage associated with an explosion using this method is as follows:
Determine the total quantity of flammable material involved in the explosion.
Estimate the explosion efficiency and calculate the equivalent mass of TNT using Equation 6-30.
Use the scaling law given by Equation 6-27 and Figure 6-24 (or Equation 6-29) to estimate the peak side-on overpressure.
Use Table 6-9 to estimate the damage for common structures and process equipment.
This procedure can be applied in reverse to estimate the quantity of material involved based on damage estimates.
TNO Multi-Energy Method
The TNO method identifies the confined volumes in a process, assigns a relative degree of confinement or congestion, and then determines the contribution to the overpressure from this confined volume (TNO is an acronym for the Netherlands Organization for Applied Scientific Research). Semi-empirical curves are used to determine the overpressure. The basis for this model is that the energy of explosion depends highly on the level of congestion and depends less on the fuel in the cloud.
The procedure for using the multienergy model for a VCE is as follows:21
Use a dispersion model to determine the extent of the cloud. In general, this is done by assuming that equipment and buildings are not present, because of the limitations of dispersion modeling in congested areas.
Conduct a field inspection to identify the congested areas. In most cases, heavy vapors tend to move downhill.
Identify potential sources of strong blasts within the area covered by the flammable cloud. Potential sources of strong blasts include congested areas and buildings, such as process equipment in chemical plants or refineries, stacks of crates or pallets, and pipe racks; spaces between extended parallel planes (e.g., those beneath closely parked cars in parking lots, as well as open buildings such as multistory parking garages); spaces within tube-like structures (e.g., tunnels, bridges, corridors, sewage systems, and culverts); and an intensely turbulent fuel–air mixture in a jet resulting from release at high pressure. The remaining fuel–air mixture in the flammable cloud is assumed to produce a blast of minor strength.
Estimate the energy of equivalent fuel–air charges by (a) considering each blast source separately; (b) assuming that the full quantities of fuel–air mixture present within the partially confined/obstructed areas and jets, identified as blast sources in the cloud, contribute to the blasts; (c) estimating the volumes of fuel–air mixture present in the individual areas identified as blast sources (this estimate can be based on the overall dimensions of the areas and jets; note that the flammable mixture may not fill an entire blast source volume and that the volume of equipment should be considered where it represents an appreciable proportion of the whole volume); and (d) calculating the combustion energy E (in joules) for each blast by multiplying the individual volumes of the mixture by 3.5 × 106 J/m3 (this value is typical for the heat of combustion of an average stoichiometric hydrocarbon–air mixture).
Assign a number representative of the blast strength for each individual blast. Some companies have defined procedures for this; however, many risk analysts use their own judgment.
A safe and most conservative estimate of the strength of the sources of a strong blast can be made if a maximum strength of 10—representative of a detonation—is assumed. However, a source strength of 7 seems to more accurately represent actual experience. Furthermore, for side-on overpressures less than approximately 0.5 bar, no differences appear for source strengths ranging from 7 to 10.
The blast resulting from the remaining unconfined and unobstructed parts of a cloud can be modeled by assuming a low initial strength. For extended and quiescent parts, assume a minimum strength of 1. For more nonquiescent parts, which are in low-intensity turbulent motion (for instance, because of the momentum of a fuel release), assume a strength of 3.
Once the energy quantities E and the initial blast strengths of the individual equivalent fuel–air charges are estimated, the Sachs-scaled blast side-on overpressure and positive-phase duration at some distance R from a blast source is read from the blast charts in Figure 6-25 after calculation of the Sachs-scaled distance:
ˉR=R(E/Po)1/3(6-31)

21Guidelines for Evaluating the Characteristics of Vapor Cloud Explosions, Flash Fires, and BLEVEs (New York, NY: American Institute of Chemical Engineers, 1994).
where
ˉR
R is the distance from the charge (m),
E is the charge combustion energy (J), and
Po is the ambient pressure (Pa).
The blast peak side-on overpressure and positive-phase duration are calculated from the Sachs-scaled overpressure and the Sachs-scaled positive-phase duration. The overpressure is given by
p0 = ΔˉPs•pa(6-32)
and the positive-phase duration is given by
td= ˉtd[(E/Pa)1/3co](6-33)
where
Po
ΔˉPs
pa is the ambient pressure (Pa),
td is the positive-phase duration (s),
ˉtd
E is the charge combustion energy (J), and
co is the ambient speed of sound (m/s).
If separate blast sources are located close to one another, they may be initiated almost simultaneously, and the respective blasts should be added together. The most conservative approach to this issue is to assume a maximum initial blast strength of 10 and to sum the combustion energy from each source in question. Further definition of this important issue (for instance, the determination of a minimum distance between potential blast sources so that their individual blasts can be considered separately) is a factor in present research.
The major problem with the application of the TNO multienergy method is that the user must decide on the selection of a severity factor, based on the degree of confinement or congestion. Little guidance is available for partial confinement geometries. Furthermore, it is not clear how the results from each blast strength should be combined.
Another popular method to estimate overpressures is the Baker–Strehlow–Tang method. This method is based on a flame speed, which is selected depending on three factors: (1) the reactivity of the released material, (2) the flame expansion characteristics of the process unit (which relates to confinement and spatial configuration), and (3) the obstacle density within the process unit. A set of semi-empirical curves is used to determine the overpressure. A complete description of the procedure is provided by Baker et al.22
22Q. A. Baker, C. M. Doolittle, G. A. Fitzgerald, and M. J. Tang. “Recent Developments in the Baker– Strehlow VCE Analysis Methodology.” Process Safety Progress 17, no. 4 (1998): 297.
The TNO multienergy and Baker–Strehlow–Tang methods are essentially equivalent, although the TNO method tends to predict a higher pressure in the near field and the Baker–Strehlow method tends to predict a higher pressure in the far field. The confinement and congestion are better defined in the Baker–Strehlow–Tang method than in the TNO method. Both methods require more information and detailed calculations than the TNT equivalency method. In particular, both require identifying regions of high confinement or congestion in the process area.
Energy of Chemical Explosions
The blast wave resulting from a chemical explosion is generated by the rapid expansion of gases at the explosion site. This expansion can be caused by two mechanisms: (1) thermal heating of the reaction products and (2) the change in the total number of moles by reaction.
For most hydrocarbon combustion explosions in air, the change in the number of moles is small. For example, consider the combustion of propane in air. The stoichiometric equation is
C3H8 + 5O2 + 18.8N2→3CO2 + 4H2O + 18.8N2
The initial number of moles on the left-hand side is 24.8, and the number of moles on the right-hand side is 25.8. In this case. only a small pressure increase is expected as a result of the change in the number of moles, and almost all the blast energy is due to thermal energy release.
The energy released during an explosion is computed using standard thermodynamics. Typically, the heat of combustion is used, but the reaction energy can be easily computed using standard heats of formation. Heat of combustion data are provided in Appendix B. In most cases, the lower heat of combustion is used, where the water product is in the vapor phase, not liquid. Since an explosion occurs within a few milliseconds, the explosive energy is released long before the water vapor can condense into liquid.
The released explosion energy is equal to the work required to expand the gases. Crowl reasoned that this expansion work is a form of mechanical energy.23 The thermodynamic availability is a state function used to determine the maximum mechanical energy extractable from a material as it moves into equilibrium with its surroundings. Sussman showed that the thermodynamic availability for a reacting system can be computed using the standard Gibbs energy of formation.24 Crowl then concluded that the energy of explosion for a material exploding at room temperature and pressure is equal to the standard Gibbs energy of formation. Crowl also showed how the energy of explosion could be determined for materials exploding at different gas compositions and nonambient temperatures and pressures. However, these adjustments are normally small.
23D. A. Crowl. “Calculating the Energy of Explosion Using Thermodynamic Availability.” Journal of Loss Prevention in the Process Industries 5, no. 2 (1992): 109–118.
24M. V. Sussman. Availability (Exergy) Analysis (Lexington, MA: Mulliken House, 1981).
Example 6-11
One thousand kilograms of methane escapes from a storage vessel, mixes with air, and explodes. Determine (a) the equivalent amount of TNT and (b) the side-on peak overpressure at a distance of 50 m from the blast. Assume an explosion efficiency of 2%.
Solution
The heat of combustion for methane is found in Appendix B. Substituting into Equation 6-30, we obtain
mTNT = ηmΔHcETNT = (0.02)(1000 kg)(1 mo1/0.016 kg)(802.3 kJ/mol)4686 kJ/kg = 214 kg TNT
mTNT = ηmΔHcETNT = (0.02)(1000 kg)(1 mo1/0.016 kg)(802.3 kJ/mol)4686 kJ/kg = 214 kg TNT Equation 6-27 is used to determine the scaled distance:
ze = rm1/3TNT = 50 m(214 kg)1/3 = 8.4 m/kg1/3
ze = rm1/3TNT = 50 m(214 kg)1/3 = 8.4 m/kg1/3 From Figure 6-24 (or Equation 6-29), the scaled overpressure is 0.25. Thus the overpressure is
p0= pspa= (0.25)(101.3 kPa)=25 kPa
p0= pspa= (0.25)(101.3 kPa)=25 kPa From Table 6-9, this overpressure is adequate to demolish steel panel buildings.
Example 6-12
Consider the explosion of a propane–air vapor cloud confined beneath a storage tank. The tank is supported 1 m off the ground by concrete piles. The concentration of vapor in the cloud is assumed to be at stoichiometric concentrations. Assume a cloud volume of 2094 m3, confined below the tank, representing the volume underneath the tank. Determine the overpressure from this vapor cloud explosion at a distance of 100 m from the blast using the TNO multienergy method.
Solution
The heat of combustion of a stoichiometric hydrocarbon–air mixture is approximately 3.5 MJ/m3, and by multiplying by the confined volume, the resulting total energy is (2094 m3)(3.5 MJ/m3) = 7329 MJ. To apply the TNO multienergy method, a blast strength of 7 is chosen. The Sachs-scaled energy is determined using Equation 6-26. The result is
ˉR=R(E/Po)1/3=100 m[(7329×106 J)/(101,325 Pa)]1/3=2.4
The curve labeled 7 in Figure 6-26 is used to determine the scaled overpressure value of about 0.13. The resulting side-on overpressure is determined from Equation 6-30:
p0 = ΔˉPs⋅pa = (0.13)(101.3 kPa) = 13.2 kPa = 1.9 psi.

From Table 6-9, it appears that this explosion will result in distortion of steel frame buildings and is almost strong enough to result in partial collapse of walls and roofs of houses.
Energy of Mechanical Explosions
For mechanical explosions, a reaction does not occur and the energy is obtained from the energy content of the contained substance. If this energy is released rapidly, an explosion may result. Examples of this type of explosion are the sudden failure of a tire full of compressed air and the sudden catastrophic rupture of a compressed gas tank.
Four methods are used to estimate the energy of explosion for a pressurized gas: Brode’s equation, isentropic expansion, isothermal expansion, and thermodynamic availability. Brode’s method25 is perhaps the simplest approach. It determines the energy required to raise the pressure of the gas at constant volume from atmospheric pressure to the gas pressure in the vessel. The resulting expression is
25H. L. Brode. “Blast Waves from a Spherical Charge.” Physics of Fluids 2 (1959): 17.
E = (P2−P1)Vγ−1(6-34)
where
E is the energy of explosion (energy),
P1 is the ambient pressure (force/area),
P2 is the burst pressure of the vessel (force/area),
V is the volume of expanding gas in the vessel (volume), and
γ is the heat capacity ratio for the gas (unitless).
Because P2 > P1, the energy calculated from Equation 6-34 is positive, indicating that the energy is released to the surroundings during the vessel rupture.
The isentropic expansion method assumes that the gas expands isentropically from its initial to final state. The following equation represents this case:
E=(P2Vγ−1)[1−(P1P2)(γ−1)/γ](6-35)
The isothermal expansion case assumes that the gas expands isothermally. This is represented by the following equation:
E=nRgT1ln(P2P1) = P2Vln(P2P1)(6-36)
where
n is the number of moles of gas (moles),
Rg is the ideal gas constant, and
T1 is the ambient temperature (degrees).
The final method uses thermodynamic availability to estimate the energy of explosion. Thermodynamic availability represents the maximum mechanical energy extractable from a material as it comes into equilibrium with the environment. The resulting overpressure from an explosion is a form of mechanical energy. Thus, thermodynamic availability predicts a maximum upper bound to the mechanical energy available to produce an overpressure.
An analysis by Crowl using batch thermodynamic availability resulted in the following expression to predict the maximum explosion energy of a gas contained within a vessel:26
26D. A. Crowl. “Calculating the Energy of Explosion Using Thermodynamic Availability.” Journal of Loss Prevention in the Process Industries 5, no. 2 (1992): 109–118.
E = P2V[ln(P2P1)−(1−P1P2)](6-37)
Note that Equation 6-37 is nearly the same as Equation 6-36 for an isothermal expansion, with the addition of a correction term. This correction term accounts for the energy lost as a result of the second law of thermodynamics.
The question arises as to which method to use. Figure 6-26 presents the energy of explosion using all four methods as a function of initial gas burst pressure of the vessel. The calculation assumes an inert gas initially at 298 K with γ=1.4. The gas expands into ambient air at 1 atm pressure. The isentropic method produces a low value for the energy of explosion. The isentropic expansion results in a gas at a very low temperature; the expansion of an ideal gas from 200 psia to 14.7 psia results in a final temperature of 254°R, or -254°F This is thermodynamically inconsistent because the final temperature is ambient. The isothermal expansion method predicts a large value for the energy of explosion because it assumes that all the energy of compression is available to perform work. In reality, some of the energy must be expelled as waste heat, according to the second law of thermodynamics. The thermodynamic availability method accounts for this loss through the correction term in Equation 6-37. All four methods continue to be used to estimate the energy of explosion for compressed gases.
It is thought that Brode’s equation more closely predicts the potential explosion energy close to the explosion source (near field), and that the isentropic expansion method predicts better the effects at a greater distance (far field). However, it is unclear where this transition occurs. Also, a portion of the potential explosion energy of vessel burst is converted into kinetic energy of the vessel pieces and other inefficiencies (such as strain energy in the form of heat in the vessel fragments).
Missile Damage
An explosion occurring in a confined vessel or structure can rupture the vessel or structure, resulting in the projection of debris over a wide area. This debris (also called missiles) can cause appreciable injury to people and damage to structures and process equipment. Moreover, such missiles can be projected to large distances from the explosion, causing additional remote damage.
Missiles are frequently a means by which an accident propagates throughout a plant facility. For example, a localized explosion in one part of the plant may project debris throughout the plant. This debris strikes storage tanks, process equipment, and pipelines, resulting in secondary fires or explosions. This chain of damage is called the “domino effect” for explosions.
Clancey developed an empirical relationship between the mass of explosive and the maximum horizontal range of the fragments,27 as illustrated in Figure 6-27. This relationship is useful during accident investigations for calculating the energy level required to project fragments over an observed distance.
27V. J. Clancey. “Diagnostic Features of Explosion Damage,” paper presented at the Sixth International Meeting of Forensic Sciences, Edinburgh, UK, 1972.

Blast Damage to People
People can be injured by explosions from direct blast effects (including overpressure and thermal radiation) or indirect blast effects (mostly missile damage). Blast damage effects are estimated using probit analysis, as discussed in Section 2-6.
Example 6-13
A reactor contains the equivalent of 5000 kg of TNT. If it explodes, estimate the injury to people and the damage to structures located 15 m away.
Solution
The overpressure is determined using Equation 6-27 and Figure 6-24. The scaled distance is
ze =rm1/3TNT =150 m(5000 kg)1/3 =8.80 m/kg1/3
From Figure 6-24, the scaled overpressure is 0.20 and the overpressure is (0.20) (101.3 kPa) = 20.3 kPa. Table 6-9 indicates that this will result in 50% destruction of brickwork of houses.
Injury to personnel is determined using probit equations from Table 2-5. The probit equation for deaths resulting from lung hemorrhage is
Y=−77.1 + 6.91 in P
and the probit equation for eardrum rupture is
Y=−15.6 + 1.93 in P
where P is the overpressure in N/m2. 20.3 kPa = 20,300 N/m2.
Substituting this value into the probit equations yields
Ydeaths = −77.1 + 6.91 ln(20,300 N/m2)=−8.56
Yeardrums = −15.6 + 1.93 ln(20,300 N/m2)=3.54
Table 2-4 converts the probit to percentages. The result shows that there should be no deaths and that fewer than 7% to 8% of the exposed people would suffer eardrum ruptures. This assumes complete conversion of explosion energy.
Vapor Cloud Explosions (VCE)
The most dangerous and destructive explosions in the chemical process industries are VCEs. These explosions occur as the following sequence of events:
Sudden release of a large quantity of flammable vapor (typically this occurs when a vessel, containing a superheated and pressurized liquid, ruptures)
- Dispersion of the vapor throughout the plant site while mixing with air
Ignition of the resulting vapor cloud
The accident at Flixborough, England, is a classic example of a VCE. The sudden failure of a 20-inch cyclohexane line between reactors led to almost instantaneous vaporization of an estimated 30 tons of cyclohexane. The vapor cloud dispersed throughout the plant site, mixed with air, and was ignited by an unknown source 45 seconds after the release. The entire plant site was leveled and 28 people were killed.
A summary of 29 international VCEs over the period 1974–1986 shows property losses for each event of between $5,000,000 and $100,000,000 and 140 fatalities (an average of almost 13 per year).28 Any process containing quantities of liquefied gases, volatile superheated liquid, or high-pressure gases is considered a good candidate for a VCE.
28Richard W. Prugh. “Evaluation of Unconfined Vapor Cloud Explosion Hazards.” In International Conference on Vapor Cloud Modeling (New York, NY: American Institute of Chemical Engineers, 1987), p. 713.
VCEs are difficult to characterize, primarily because of the large number of parameters needed to describe an event. Some of the parameters that affect VCE behavior are quantity of material released, fraction of material vaporized, probability of ignition of the cloud, distance traveled by the cloud before ignition, time delay before ignition of cloud, probability of explosion rather than fire, existence of a threshold quantity of material, efficiency of explosion, and location of ignition source with respect to release.29 Accidents occur under uncontrolled circumstances, and data collected from real events are difficult to quantify and compare.
29Sam Mannan, ed. Lees’ Loss Prevention in the Process Industries, 3rd ed. (Amsterdam, Netherlands: Elsevier, 2005), p. 17/134.
Qualitative studies30 have shown that (1) the ignition probability increases as the size of the vapor cloud increases; (2) vapor cloud fires are more common than explosions; (3) the explosion efficiency is usually small (approximately 2% of the combustion energy is converted into a blast wave); and (4) turbulent mixing of vapor and air and ignition of the cloud at a point remote from the release increases the impact of the explosion.31 Congestion due to process equipment or confinement is also an important issue.
30Sam Mannan, ed. Lees’ Loss Prevention in the Process Industries, 3rd ed. (Amsterdam, Netherlands: Elsevier, 2005), p. 17/140.
31Richard W. Prugh. “Evaluation of Unconfined Vapor Cloud Explosion Hazards.” In International Conference on Vapor Cloud Modeling (New York, NY: American Institute of Chemical Engineers, 1987), p. 714.
From a safety standpoint, the best approach is to prevent the release of material. A large cloud of combustible material is hazardous and almost impossible to control, despite any safety systems installed to prevent ignition. Methods that are used to prevent VCEs include keeping low inventories of volatile and flammable materials, using process conditions that minimize flashing if a vessel or pipeline is ruptured, using analyzers to detect leaks at low concentrations, and installing automated block valves to shut systems down while the spill or release is in the incipient stage of development.
Boiling-Liquid Expanding-Vapor Explosions32
32Sam Mannan, ed. Lees’ Loss Prevention in the Process Industries, 3rd ed. (Amsterdam, Netherlands: Elsevier, 2005), p. 17/167; Frank Bodurtha. Industrial Explosion Prevention and Protection (New York, NY: McGraw- Hill, 1980), p. 99.
A boiling-liquid expanding-vapor explosion (BLEVE, pronounced ble’-vee) is a special type of accident that can explosively release large quantities of materials. It occurs when a tank containing a liquid held above its atmospheric pressure boiling point ruptures, resulting in the explosive vaporization of a large fraction of the tank contents. If the materials are flammable, a VCE might result; if they are toxic, a large area might be subjected to toxic materials. In either situation, the energy released by the BLEVE process itself can result in considerable damage.
BLEVEs are caused by the sudden failure of the container as a result of any cause. The most common type of BLEVE is caused by external fire. The steps resulting in this type of event are as follows:
A fire develops external and adjacent to a tank containing a liquid.
The fire heats the walls of the tank.
The tank walls below liquid level are cooled by the liquid, increasing the liquid temperature and the pressure in the tank.
If the flames reach the tank walls or roof where there is only vapor and no liquid to remove the heat, the tank metal temperature rises until the tank loses it structural strength.
The tank ruptures, explosively vaporizing its contents.
It is likely that the vessel will fail at a pressure below the design pressure of the vessel due to the weakening of the vessel walls due to the thermal heating. It is also likely that the vessel will fail below the set pressure of any pressure protection systems on the vessel.
If the liquid is flammable and a fire is the cause of the BLEVE, the liquid will ignite as the tank ruptures. Often, the boiling and burning liquid behaves as a rocket fuel, propelling vessel parts for great distances. When a BLEVE occurs in a vessel, only a fraction of the liquid vaporizes; the amount depends on the physical and thermodynamic conditions of the vessel contents. The fraction vaporized is estimated using the methods discussed in Section 4-7.
Suggested Reading
Center for Chemical Process Safety. Guidelines for Evaluating Process Buildings for External Explosions and Fires (New York, NY: John Wiley, 1996).
Center for Chemical Process Safety. Guidelines for Vapor Cloud Explosion, Pressure Vessel Burst, BLEVE and Flash Fire Hazards, 2nd ed. (New York, NY: John Wiley, 2010).
D. A. Crowl. Understanding Explosions (New York, NY: John Wiley, 2003).
Rolf Eckhoff. Dust Explosions in the Process Industries, 3rd ed. (Amsterdam, Netherlands: Elsevier, 2003).
Rolf Eckhoff. Explosion Hazards in the Process Industries, 2nd ed. (Cambridge, MA: Gulf Professional Publishing, 2016).
Irvin Glassman and Richard A. Yetter. Combustion, 4th ed. (Burlington, MA: Academic Press, 2008).
Don W. Green and Robert H. Perry, eds. Perry’s Chemical Engineers’ Handbook, 9th ed. (New York, NY: McGraw-Hill, 2019), pp. 23–6 to 23–18.
Gilbert F. Kinney and Kenneth J. Graham. Explosive Shocks in Air, 2nd ed. (Berlin, Germany: Springer-Verlag, 1985).
Bernard Lewis and Guenther von Elbe. Combustion, Flames, and Explosions of Gases, 3rd ed. (Burlington, MA: Academic Press, 1987).
Sam Mannan, ed. Lees’ Loss Prevention in the Process Industries, 3rd ed. (Amsterdam, Netherlands: Elsevier, 2005), ch. 16 and 17.
Society of Fire Protection Engineers. SFPE Handbook of Fire Protection Engineering, 5th ed. (Quincy, MA: National Fire Protection Association, 2015).
Daniel R. Stull. Fundamentals of Fires and Explosion, AICHE Monograph Series, no. 10, v. 73 (New York, NY: American Institute of Chemical Engineers, 1977).
Problems
6-1. What is the flash point temperature (in °C) of a 50 mole percent mixture of methanol and water? Use Appendix B for the flash point temperature for pure methanol and Appendix C for Antoine equations for vapor pressure.
6-2. A gas mixture is composed of 15% carbon monoxide, 10% methane, and the rest air. Is this mixture flammable? Use the flammability data provided in Appendix B.
6-3. Estimate the upper and lower flammable limits for carbon monoxide and heptane using the stoichiometric method via Equations 6-10 and 6-11 in the text. Compare to experimental values provided in Appendix B.
6-4. A gas cylinder contains a gas mixture composed of 50% methane and 50% ethylene by volume. Estimate the LFL and the UFL for this gas mixture. Compare to the experimental values of 3.6% for the LFL and 21.5% for the UFL.
6-5. Estimate the UFL and the LFL for ethylene using the stoichiometric concentrations and Equations 6-10 and 6-11 in the textbook. Compare to the experimental values in Appendix B.
6-6. Estimate the LOC of ethylene using Equations 6-15 and 6-16 in the textbook. Compare to the experimental value in Table 6-3.
6-7. Estimate the LOC for (a) carbon monoxide and (b) heptane using Equation 6-15. Compare to experimental values in Table 6-3.
6-8. Draw an approximate flammability triangle diagram for methyl alcohol. Use published flammability data from Appendix B and Table 6-3. If a gas containing 20% methyl alcohol, 5% oxygen, and 75% nitrogen is mixed with air, will it become flammable?
6-9. A barbeque gas cylinder contains 20 lb of propane. The cylinder accidentally falls over and ruptures, vaporizing the entire contents of the cylinder. The vapor cloud is ignited and an explosion occurs. Estimate the overpressure from this explosion 100 ft away. Which type of damage is expected?
6-10. A VCE with methane destroyed a house structure 100 ft away from the ignition source. Estimate the amount of methane released.
6-11. One compartment in a gasoline tank truck has a volume of 11,860 liters.
How much gasoline liquid (in liters) is required to result in a stoichiometric concentration of gasoline vapor in this compartment?
What is the TNT equivalence of this gasoline vapor, based on a confined explosion?
Assume 1 atm and 25°C ambient conditions.
Assume that the gasoline is mostly octane (C8H18) and has the following properties:
Molecular weight of gasoline vapor: 72
Gasoline liquid specific gravity: 0.72
Lower heating value of gasoline: 44 MJ/kg
6-12. The LFL for gasoline is 1.3 vol. % gasoline vapor in air. What evaporation rate of gasoline vapor (in kg/s) will result in a downwind concentration of one-fourth of the LFL at 0.1 km directly downwind from the release?
Assume:
Worst-case stability conditions
1 atm and 25°C ambient conditions
The same physical properties for gasoline provided in Problem 6-11
6-13. Liquid heptane is stored in a 100,000-L storage vessel that is vented directly to air. The heptane is stored at 25°C and 1 atm pressure. The liquid is drained from the storage vessel and all that remains in the vessel is the air saturated with heptane vapor.
Is the vapor in the storage vessel flammable?
What is the TNT equivalent for the vapor remaining in the vessel?
If the vapor explodes, what is the overpressure 50 m from the vessel?
What damage can be expected at 50 m?
Data for heptane (C7H16):
Molecular weight: 100.21
Lower heat of combustion: –4464.9 kJ/mol
LFL:1.0 vol. %
UFL: 7.0 vol. %
Boiling point: 98.4°C
6-14. A plant is considering installing a butane storage sphere to be located 100 m from the control room building. You are asked to review the consequences of a vapor cloud explosion involving all of the sphere’s inventory of butane. The control room building is designed only for a 3 psi (20.7 kPa) overpressure. What is the maximum quantity of butane (in kg) that can be stored without exceeding the 3 psi pressure specification?
Data for butane: LHV of –2657.5 kJ/mol (from Appendix B)
6-15. A set of experiments is run on a flammable gas in a spherical vessel. The following data are obtained for two different vessel volumes. Estimate the value of KG for this combustible gas:
V = 1 m3 |
V = 20 m3 |
||
|---|---|---|---|
Time (s) |
P (bar) |
Time (s) |
P (bar) |
0.0 |
0.0 |
0.0 |
0.0 |
0.1 |
0.2 |
0.2 |
0.15 |
0.2 |
0.5 |
0.3 |
0.35 |
0.3 |
1.2 |
0.4 |
0.6 |
0.35 |
1.6 |
0.5 |
0.9 |
0.40 |
3.2 |
0.6 |
1.4 |
0.425 |
4.7 |
0.7 |
2.2 |
0.450 |
6.5 |
0.8 |
4.1 |
0.475 |
6.9 |
0.85 |
5.0 |
0.500 |
7.1 |
0.90 |
6.2 |
0.550 |
7.4 |
0.95 |
7.1 |
0.600 |
7.3 |
1.00 |
7.0 |
0.650 |
7.0 |
1.05 |
7.2 |
0.700 |
6.4 |
1.10 |
6.7 |
0.750 |
6.1 |
1.15 |
6.25 |
0.800 |
5.7 |
1.20 |
5.90 |
0.900 |
5.1 |
1.30 |
5.40 |
1.000 |
4.7 |
1.40 |
5.00 |
|
|
1.50 |
5.60 |
6-16. The following liquids are stored in storage vessels at 1 atm and 25°C. The vessels are vented with air. Determine whether the equilibrium vapor above the liquid will be flammable.
Acetone
Benzene
Cyclohexane
Ethyl alcohol
Heptane
Hexane
Pentane
Toluene
6-17. Black powder has been used as a blasting agent and a rifle propellant for hundreds, if not thousands, of years. It is a mixture of potassium nitrate, charcoal, and sulfur. The reaction that occurs during black powder combustion is
2KNO3+3C+S→K2S+3CO2+N2
Estimate the TNT equivalence (in kg) of 1 kg of black powder. The heats of formation are given below:
Species |
Heat of formation (kJ/mol) |
|---|---|
KNO3 |
–494.6 |
CO2 |
–393.6 |
K2S |
–380.7 |
Additional homework problems are available in the Pearson Instructor Resource Center.