
3.5. Explosion Limits and Oxidation Characteristics of Hydrocarbons
To establish the importance of the high-temperature chain mechanism through the H2–O2 sequence, the oxidation of H2 was discussed in detail. Also, because CO conversion to CO2 is the highly exothermic portion of any hydrocarbon oxidation system, CO oxidation was then detailed. Since it will be shown that all carbon atoms in alkyl hydrocarbons and most in aromatics are converted to CO through the radical of formaldehyde (H2CO) called formyl (HCO), the oxidation of aldehydes will be the next species to be considered. Then the sequence of oxidation reactions of the C1 to C5 alkyl hydrocarbons is considered. These systems provide the backdrop for consideration of the oxidation of the hydrocarbon oxygenates—alcohols, ether, ketenes, etc. The oxidation of the highly stabilized aromatics is then analyzed. Finally, a brief discussion of the oxidation of biofuels is provided. This hierarchical approach should facilitate the understanding of the oxidation of most hydrocarbon fuels.
The approach is to start with analysis of the smallest of the hydrocarbon molecules, methane. It is interesting that the combustion mechanism of methane was for a long period of time the least understood. In recent years, however, there have been many studies of methane so that to a large degree its specific oxidation mechanisms are known over various ranges of temperatures. Now among the best understood, these mechanisms will be detailed later in this chapter.
The higher-order hydrocarbons, particularly propane and above, oxidize much more slowly than hydrogen and are known to form metastable molecules that are important in explaining the explosion limits of hydrogen and carbon monoxide. The existence of these metastable molecules makes it possible to explain qualitatively the unique explosion limits of the complex hydrocarbons and to gain some insights into what the oxidation mechanisms are likely to be.
Mixtures of hydrocarbons and oxygen react very slowly at temperatures below 200 °C; as the temperature increases, a variety of oxygen-containing compounds can begin to form. As the temperature is increased further, CO and H2O begin to predominate in the products and H2O2 (hydrogen peroxide), CH2O (formaldehyde), CO2, and other compounds begin to appear. At 300–400 °C, a faint light often appears, and this light may be followed by one or more blue flames that successively traverse the reaction vessel. These light emissions are called cool flames and can be followed by an explosion. Generally, the presence of aldehydes is revealed.
In discussing the mechanisms of hydrocarbon oxidation and, later, in reviewing the chemical reactions in photochemical smog, it becomes necessary to identify compounds whose structure and nomenclature may seem complicated to those not familiar with organic chemistry. One need not have a background in organic chemistry to follow the combustion mechanisms; one should, however, study the following section to obtain an elementary knowledge of organic nomenclature and structure.
3.5.1. Organic Nomenclature
No attempt is made to cover all the complex organic compounds that exist. The classes of organic compounds reviewed are those that occur most frequently in combustion processes and photochemical smog.
3.5.1.1. Alkyl compounds
| Paraffins (alkanes: single bonds)  | CH4, C2H6, C3H8, C4H10,…, CnH2n+2 methane, ethane, propane, butane,…, straight chain; isobutane, branched chain All are saturated (i.e., no more hydrogen can be added to any of the compounds) Radicals deficient in one H atom take the names methyl, ethyl, propyl, etc. |
| Olefins (alkenes: contain double bonds)  | C2H4, C3H6, C4H8, …, CnH2n ethene, propene, butene (ethylene, propylene, butylene) Diolefins contain two double bonds The compounds are unsaturated, since CnH2n can be saturated to CnH2n+2 |
| Table Continued | |

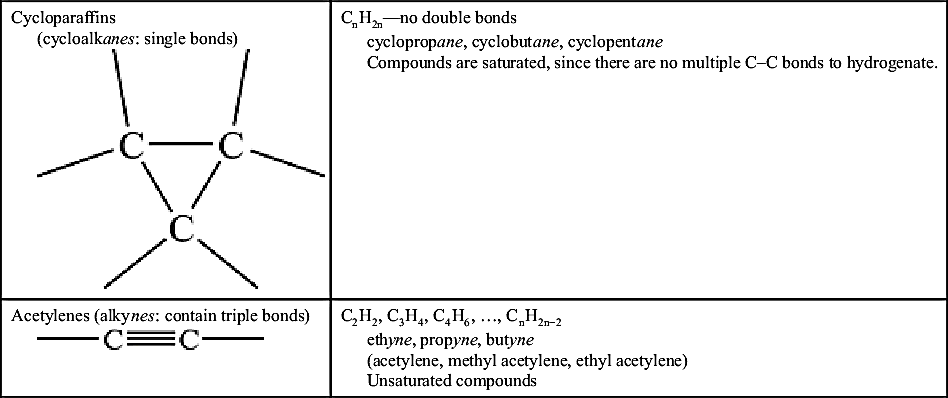
| Cycloparaffins (cycloalkanes: single bonds)  | CnH2n—no double bonds cyclopropane, cyclobutane, cyclopentane Compounds are saturated, since there are no multiple C–C bonds to hydrogenate. |
| Acetylenes (alkynes: contain triple bonds) | C2H2, C3H4, C4H6, …, CnH2n−2 ethyne, propyne, butyne (acetylene, methyl acetylene, ethyl acetylene) Unsaturated compounds |
3.5.1.2. Aromatic compounds
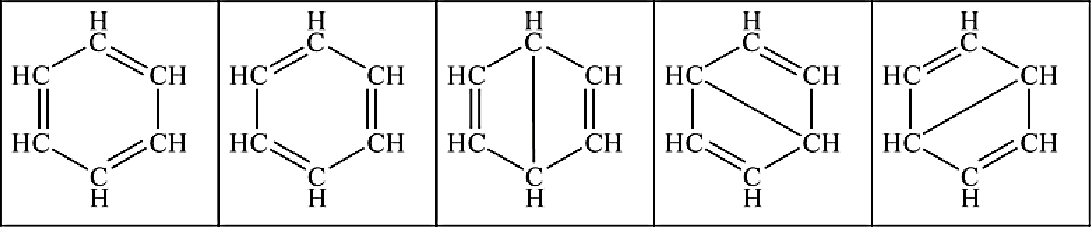
The building block for the aromatics is the ring-structured benzene C6H6, which has many resonance structures and is therefore very stable:
 |  |  |  |  |
The ring structure of benzene is written in shorthand as either
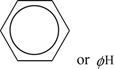
where ϕ is the phenyl radical: C6H5. Thus
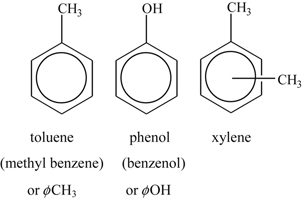
xylene being ortho, meta, or para according to whether methyl groups are separated by one, two, or three carbon atoms, respectively (which are also referred to as 1,2-dimethylbenzene, 1,3-dimethylbenzene, and 1,4-dimethylbenzene, respectively).
Polycyclic aromatic hydrocarbons (PAH) are those which there exist combined aromatic ring structures represented by naphthalene (C10H8);

α-methylnaphthalene (or 1-methylnaphthalene) has a methyl radical attachment at one of the peak carbon atoms (i.e., a carbon atom adjacent to the central two). If β is used (also referred to as 2-methylnaphthalene), than the methyl radical is attached to one of the other nonassociated carbon atoms.
3.5.1.3. Alcohols
Those organic compounds that contain a hydroxyl group (–OH) are called alcohols and follow the simple naming procedure.
![]()
The bonding arrangement is always

3.5.1.4. Aldehydes
The aldehydes contain the characteristic formyl radical group

and can be written as


3.5.1.5. Ketones
The ketones contain the characteristic group

and can be written more generally as
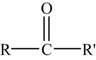
where R and R′ are always organic radicals. Thus
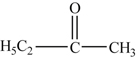
is methyl ethyl ketone (also known as butanone).
3.5.1.6. Organic acids
Organic acids contain the group

and are generally written as
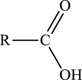
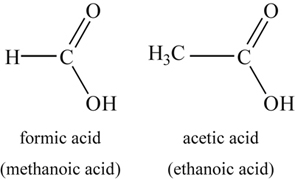
3.5.1.7. Organic salts

3.5.1.8. Other
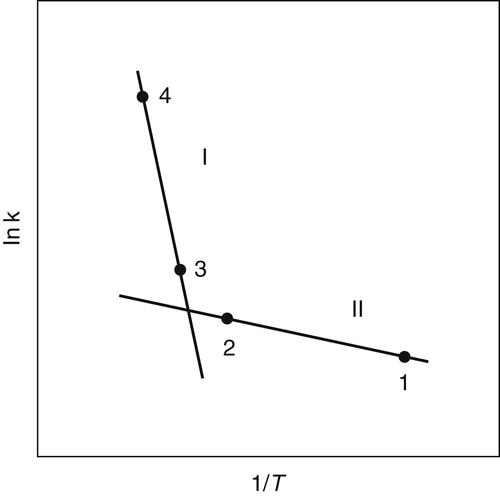
Ethers take the form 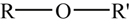 , where R and R′ are organic radicals. The peroxides take the form
, where R and R′ are organic radicals. The peroxides take the form  or
or  , in which case the term hydroperoxide is used. The functional group C=O that appears in the aldehydes, ketones, and organic (carboxylic) acids above is referred to the carbonyl group. An ester consists of a carbonyl group adjacent to an ether linkage, that is,
, in which case the term hydroperoxide is used. The functional group C=O that appears in the aldehydes, ketones, and organic (carboxylic) acids above is referred to the carbonyl group. An ester consists of a carbonyl group adjacent to an ether linkage, that is,

3.5.2. Explosion Limits
At temperatures around 300–400 °C and slightly higher, explosive reactions in hydrocarbon–air mixtures can take place. Thus, explosion limits exist in hydrocarbon oxidation. A general representation of the explosion limits of hydrocarbons is shown in Figure 3.9.
The shift of curves, as shown in Figure 3.9, is unsurprising, since the larger fuel molecules and their intermediates tend to break down more readily to form radicals that initiate fast reactions. The shape of the propane curve suggests that branched chain mechanisms are possible for hydrocarbons. One can conclude that the character of the propane mechanism is different from that of the H2–O2 reaction when one compares this explosion curve with the H2–O2 pressure peninsula. The island in the propane–air curve drops and goes slightly to the left for higher-order paraffins; for example, for hexane it occurs at 1 atm. For the reaction of propane with pure oxygen, the curve drops to about 0.5 atm.

Hydrocarbons exhibit certain experimental combustion characteristics that are consistent both with the explosion limit curves and with practical considerations; these characteristics are worth reviewing:
• Hydrocarbons exhibit induction intervals that are followed by a very rapid reaction rate. Below 400 °C, these rates are of the order of 1 s or a fraction thereof, and below 300 °C they are of the order of 60 s.
• Their rate of reaction is inhibited strongly by adding surface (therefore, an important part of the reaction mechanism must be of the free-radical type).
• They form aldehyde groups, which appear to have an influence (formaldehyde is the strongest). These groups accelerate and shorten the ignition lags.
• They exhibit cool flames.
• They exhibit negative temperature coefficients of reaction rate.
• They exhibit two-stage ignition, which may be related to the cool-flame phenomenon.
• Their reactions are explosive without appreciable self-heating (branched chain explosion without steady temperature rise). Explosion usually occurs when passing from region 1 to region 2 in Figure 3.9. Explosions may occur in other regions as well, but the reactions are so fast that one cannot tell whether they are self-heating or not.
3.5.2.1. The negative coefficient of reaction rate
Semenov [16] explained the long induction period by hypothesizing unstable, but long-lived species that form as intermediates and then undergo different reactions according to the temperature. This concept can be represented in the form of the following competing fuel (A) reaction routes after the formation of the unstable intermediate M∗:

Route I is controlled by an activation energy process larger than that of II.
Figure 3.10 shows the variation of the reaction rate of each step as a function of temperature. The numbers in Figure 3.10 correspond to the temperature position designation in Figure 3.9. At point 1 in Figure 3.10, one has a chain branching system, since the temperature is low and αcrit is large; thus, α < αcrit and the system is nonexplosive. As the temperature is increased (point 2), the rate constants of the chain steps in the system increase and αcrit drops; so α > αcrit and the system explodes. At a still higher temperature (point 3), the non-chain branching route I becomes faster. Although this step is faster, α is always less than αcrit; thus the system cannot explode. Raising temperatures along route I still further leads to a reaction so fast that it becomes self-heating and hence explosive again (point 4).
The temperature domination explains the peninsula in the P–T diagram (Figure 3.9), and the negative coefficient of reaction rate is due to the shift from point 2 to 3.
3.5.2.2. Cool flames
The cool-flame phenomenon [17] is generally a result of the type of experiment performed to determine the explosion limits and the negative temperature coefficient feature of the explosion limits. The chemical mechanisms used to explain these phenomena are now usually referred to as cool-flame chemistry.
Most explosion limit experiments are performed in vessels immersed in isothermal liquid baths (see Figure 3.1). Such systems are considered to be isothermal within the vessel itself. However, the cool gases that must enter will become hotter at the walls than in the center. The reaction starts at the walls and then propagates to the center of the vessel. The initial reaction volume, which is the hypothetical outermost shell of gases in the vessel, reaches an explosive condition (point 2). However, owing to the exothermicity of the reaction, the shell's temperature rises and moves the reacting system to the steady condition point 3, and because the reaction is slow at this condition, not all the reactants are consumed. Each successive inner (shell) zone is initiated by the previous zone and progresses through the steady reaction phase in the same manner. Since some chemiluminescence occurs during the initial reaction stages, it appears as if a flame propagates through the mixture. Indeed, the events that occur meet all the requirements of an ordinary flame, except that the reacting mixture loses its explosive characteristic. Thus, there is no chance for the mixture to react completely and reach its adiabatic flame temperature. The reactions in the system are exothermic and the temperatures are known to rise about 200°C—hence the name “cool flames.”
After the complete vessel moves into the slightly higher temperature zone, it begins to be cooled by the liquid bath. The mixture temperature drops, the system at the wall can move into the explosive regime again, and the phenomenon can repeat itself since all the reactants have not been consumed. Depending on the specific experimental conditions and mixtures under study, as many as five cool flames have been known to propagate through a given single mixture. Cool flames have been observed in flow systems, as well [18].
3.5.3. “Low-Temperature” Hydrocarbon Oxidation Mechanisms
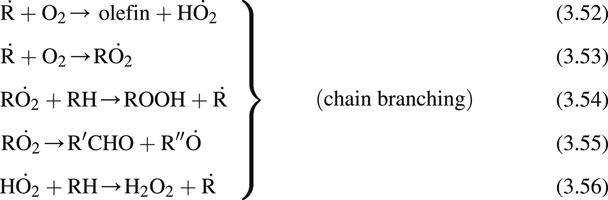
It is essential to establish the specific mechanisms that explain the cool-flame phenomenon as well as the hydrocarbon combustion characteristics mentioned earlier. Semenov [16] was the first to propose the general mechanism that formed the basis of later research, which clarified the processes taking place. This mechanism is written as follows:
![]() (3.51)
(3.51)
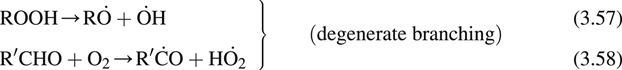
![]() (3.59)
(3.59)
where the dot above a particular atom designates the radical position. This scheme is sufficient for all hydrocarbons with a few carbon atoms, but for multicarbon (>5) species, other intermediate steps must be added, as will be shown later.
Since the system requires the buildup of ROOH (an alkyl hydroperoxide) and  before chain branching occurs to a sufficient degree to dominate the system, Semenov termed these steps degenerate branching. This buildup time, indeed, appears to account for the experimental induction times noted in hydrocarbon combustion systems. It is important to emphasize that this mechanism is a low-temperature scheme and consequently does not include the high-temperature H2–O2 chain branching steps.
before chain branching occurs to a sufficient degree to dominate the system, Semenov termed these steps degenerate branching. This buildup time, indeed, appears to account for the experimental induction times noted in hydrocarbon combustion systems. It is important to emphasize that this mechanism is a low-temperature scheme and consequently does not include the high-temperature H2–O2 chain branching steps.
At first, the question of the relative importance of ROOH versus aldehydes as intermediates was much debated; however, recent work indicates that the hydroperoxide step dominates. Aldehydes are quite important as fuels in the cool-flame region, but they do not lead to the important degenerate chain branching step as readily. The  compounds form ROH species, which play no role with respect to the branching of concern.
compounds form ROH species, which play no role with respect to the branching of concern.
Owing to its high endothermicity, the chain initiating reaction is not an important route to formation of the radical  once the reaction system has created other radicals. Obviously, the important generation step is a radical attack on the fuel, and the fastest rate of attack is by the hydroxyl radicals, since this reaction step is highly exothermic owing to the creation of water as a product. So the system for obtaining
once the reaction system has created other radicals. Obviously, the important generation step is a radical attack on the fuel, and the fastest rate of attack is by the hydroxyl radicals, since this reaction step is highly exothermic owing to the creation of water as a product. So the system for obtaining  comes from the reactions
comes from the reactions
![]() (3.60)
(3.60)
![]() (3.61)
(3.61)
where  represents any radical. It is the fate of the hydrocarbon radical that determines the existence of the negative temperature coefficient and cool flames. The alkylperoxy radical
represents any radical. It is the fate of the hydrocarbon radical that determines the existence of the negative temperature coefficient and cool flames. The alkylperoxy radical 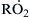 forms via reaction (3.53). The structure of this radical can be quite important. The H abstracted from RH to form the radical
forms via reaction (3.53). The structure of this radical can be quite important. The H abstracted from RH to form the radical 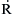 comes from a preferential position. The weakest C–H bonding is on a tertiary carbon, and if such C atoms exist, the O2 will preferentially attack this position. If no tertiary carbon atoms exist, the preferential attack occurs on the next weakest C–H bonds, which are those on the second carbon atoms from the ends of the chain. (Refer to Appendix D for all bond strengths.) Then, as the hydroxyl radical pool builds,
comes from a preferential position. The weakest C–H bonding is on a tertiary carbon, and if such C atoms exist, the O2 will preferentially attack this position. If no tertiary carbon atoms exist, the preferential attack occurs on the next weakest C–H bonds, which are those on the second carbon atoms from the ends of the chain. (Refer to Appendix D for all bond strengths.) Then, as the hydroxyl radical pool builds,  becomes the predominant attacker of the fuel. Because of the energetics of the hydroxyl step (3.61), for all intents and purposes, it is relatively nonselective in hydrogen abstraction.
becomes the predominant attacker of the fuel. Because of the energetics of the hydroxyl step (3.61), for all intents and purposes, it is relatively nonselective in hydrogen abstraction.
It is known that when O2 attaches to the radical, it forms a near 90° angle with the carbon atoms. (The realization of this steric condition will facilitate understanding of certain reactions to be depicted later.) The peroxy radical abstracts an H from any fuel molecule or other hydrogen donor to form the hydroperoxide (ROOH) (reaction (3.54)). Tracing the steps, one realizes that the amount of hydroperoxy radical that will form depends on the competition of reaction (3.53) with reaction (3.52), which forms the stable olefin together with 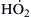 . The that forms from reaction (3.52) then forms the peroxide H2O2 through reaction (3.56). At high temperatures, H2O2 dissociates into two hydroxyl radicals; however, at the temperatures of concern here, this dissociation does not occur, and the fate of the H2O2 (usually heterogeneous) is to form water and oxygen. Thus, reaction (3.52) essentially leads only to steady reaction. In brief, then, under low-temperature conditions it is the competition between reactions (3.52) and (3.53) that determines whether the fuel–air mixture will become explosive or not. Its capacity to explode depends on whether the chain system formed is sufficiently branching to have an α greater than αcrit.
. The that forms from reaction (3.52) then forms the peroxide H2O2 through reaction (3.56). At high temperatures, H2O2 dissociates into two hydroxyl radicals; however, at the temperatures of concern here, this dissociation does not occur, and the fate of the H2O2 (usually heterogeneous) is to form water and oxygen. Thus, reaction (3.52) essentially leads only to steady reaction. In brief, then, under low-temperature conditions it is the competition between reactions (3.52) and (3.53) that determines whether the fuel–air mixture will become explosive or not. Its capacity to explode depends on whether the chain system formed is sufficiently branching to have an α greater than αcrit.
3.5.3.1. Competition between chain branching and steady reaction steps
Whether the sequence given as reactions (3.51)–(3.59) becomes chain branching or not depends on the competition between the reactions
![]() (3.52)
(3.52)
and
![]() (3.53)
(3.53)
Some evidence [19,20] suggests that both sets of products develop from a complex via a process that can be written as
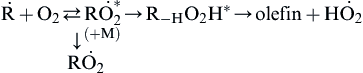 (3.62)
(3.62)
At low temperatures and modest pressures, a significant fraction of the complex dissociates back to reactants. A small fraction of the complex at low pressures then undergoes the isomerization
![]() (3.63)
(3.63)
and subsequent dissociation to the olefin and HO2. Another small fraction is stabilized to form 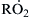 :
:
![]() (3.64)
(3.64)
With increasing pressure, the fraction of the activated complex that is stabilized will approach unity [19]. As the temperature increases, the route to the olefin becomes favored. The direct abstraction leading to the olefin reaction (3.52) must therefore become important at some temperature higher than 1000 K [20].
3.5.3.2. Importance of isomerization in large hydrocarbon radicals
With large hydrocarbon molecules an important isomerization reaction will occur. Benson [21] has noted that with six or more carbon atoms, this reaction becomes a dominant feature in the chain mechanism. Since most practical fuels contain large paraffinic molecules, one can generalize the new competitive mechanisms as
 (3.65)
(3.65)Note that the isomerization step is
![]() (3.66)
(3.66)
while the general sequence of step (d) is
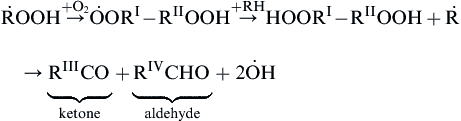 (3.67)
(3.67)
where the Roman numeral superscripts represent different hydrocarbon radicals R of smaller chain length than RH. It is this isomerization concept that requires one to add reactions to the Semenov mechanism to make this mechanism most general.
The oxidation reactions of 2-methylpentane provide a good example of how the hydroperoxy states are formed and why molecular structure is important in establishing a mechanism. The C–C bond angles in hydrocarbons are about 108°. The reaction scheme is then
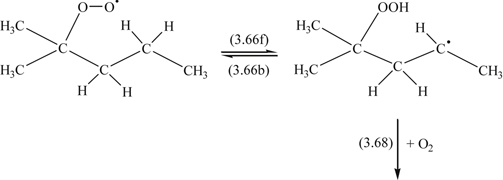 (3.63)
(3.63) (3.64)
(3.64)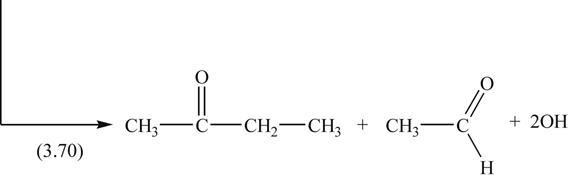 (3.65)
(3.65)Here one notices that the structure of the 90° (COO) bonding determines the intermediate ketone, aldehyde, and hydroxyl radicals that form.
Although reaction (3.66f) is endothermic and its reverse step reaction (3.66b) is faster, the competing step reaction (3.68) can be faster still; thus the isomerization step (reaction (3.66f)) controls the overall rate of formation of  and subsequent chain branching. This sequence essentially negates the extent of reaction (3.53b). Thus, the competition between
and subsequent chain branching. This sequence essentially negates the extent of reaction (3.53b). Thus, the competition between  and olefin production becomes more severe, and it is more likely that
and olefin production becomes more severe, and it is more likely that 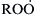 would form at the higher temperatures.
would form at the higher temperatures.
Considerable theoretical and experimental attention has been given to the energies of the R + O2 addition/dissociation reactions and their variations as the size and structure of the alkyl radical change [22–27], and recent reviews of the kinetics of elementary reactions and mechanisms of the low-temperature autoignition chemistry of hydrocarbons have appeared [28,29]. Figure 3.11 summaries the low-temperature chemistry of large hydrocarbons. In this figure, the hydroperoxyalkyl radical, that is, the product of the internal H atom transfer reaction (3.66) is called the “QOOH” species, which is common to the literature today. As discussed, each cycle of the chain consists of

1. hydrogen abstraction from the fuel by hydroxyl radicals to form the alkyl radical,
2. oxygen addition to the alkyl radical to produce the alkylperoxy radical,
3. isomerization of the alkylperoxy radical to produce the hydroperoxyalkyl radical,
4. addition of another oxygen to the hydroperoxyalkyl radical, and
Interruption of the cycle occurs when the temperature increases to a point where the alkylperoxy radical begins to dissociate back to reactants, and the alkyl radical continues to react via step (3.52) or thermally decomposes.
The greater tendency for long-chain hydrocarbons to knock as compared to smaller- and branched-chain molecules is believed to be a result of the internal isomerization branching mechanism [30]. Fuels that undergo the low-temperature chemistry of Figure 3.11 can release a significant amount of heat early on during the reaction sequence to rapidly raise the temperature of the mixture by as much as 200 K, thus shortening the time to reach the high-temperature ignition criteria discussed below.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.