
Density functional computations and mass spectrometric measurements. Can this coupling enlarge the knowledge of gas-phase chemistry?
T. Marino; N. Russo; E. Sicilia; M. Toscano Dipartimento di Chimica, Universita’ della Calabria, I-87030 Arcavacata di Rende (CS), Italy
T. Mineva Institute of Catalysis, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria
Abstract
A series of gas-phase properties of the systems has been investigated by using different exchange-correlation potentials and basis sets of increasing size in the framework of Density Functional theory with the aim to determine a strategy able to give reliable results with reasonable computational efforts.
1 Introduction
The last decade has known an extraordinary and unexpected increase of the use of Density Functional Theory (DFT) in a wide variety of chemical fields ranging from material science to homogeneous and heterogeneous catalysis, to organic, organometallic and inorganic chemistry, to biochemical and pharmacological problems [1-10]. This occurred when the traditional computational techniques (Coupled-Cluster, Multireference Configuration-Interaction, and Generalized Valence Bond approaches) resulted in great advances in their accuracy. The appeal of DFT stays in the fact that it accounts for electron correlation with a computational effort of the same order as the simplest Hartree-Fock (HF) methods.
The main characteristic of DFT is to consider the electron density as the principal variable rather than the wave function. The reliability of this theory depends on the goodness of the functionals used to describe the exchange-correlation energy.
The availability of many exchange-correlation functionals derived by different approximations (gradient-corrected, adiabatic connection), together with the possibility to express the derivatives analytically, allows to DFT to reach an high accuracy in the description of electronic and spectroscopic features of the molecular systems. This is well documented in a series of recent reviews and books [1-91].
As suggested by Kohn, Becke and Parr [11], the use of DFT is preferable (over the traditional methods) for systems with more than 5-10 atoms for which a lower accuracy is acceptable. This does not mean that for system with a less number of atoms the DFT is not reliable. With regard to this fact we mention the recent work of Seminario [6], in which, the DF atomization energy of water, obtained with different exchange-correlation functionals, is well reproduced: the results are closer to the experiment than that obtained at CCDS(T) high level of theory which is often chosen as reference when the experimental data are not available.
Despite the large number of DFT applications, relatively few works have been devoted to the reproduction or prediction of many gas-phase thermochemical properties [12-23] that are currently obtained with the modern mass-spectrometric techniques.
In this work we will show the potentiality of DFT in obtaining gas-phase chemical properties such as the proton affinity (PA), the gas-phase basicity (GPB) and acidity (GPA) and the metal ion affinity (MIA).
In addition we will consider the possibility to obtain reliable theoretical information on the preferred attach sites for proton and metal cations and on the potential energy surfaces (PES) that cannot be determined experimentally even with the most modern and sophisticated mass-spectrometric instruments [22, 23]. Furthermore, we will propose the way to rationalize some of chemical properties by using the concepts of hardness, softness and other reactivity indices (Fukui functions) for which an exact definition exists only in the framework of DFT [11]. These last fascinating tools can contribute to increase furthermore the DFT use going in the “core” of molecules to predict and explain basic chemical concepts.
2 Theoretical background
The implementation of density functional theory is based on solving the nonrelativistic Kohn-Sham one-electron equations [24] which differ from the HartreeFock ones by the inclusion of the exchange-correlation potential υxc(r). and must be solved self-consistently:



The ground state energy is given by

where εj and ρ are the self consistent quantities and Exc[ρ] is the exchange-correlation energy functional which is unknown. So, for the practical use of this theory, a good approximation for Exc[ρ] is necessary. The most widely used exchange-correlation functional employs the so-called local density approximation (LDA) [25]. In the LDA the exchange-correlation energy is given by
LDA or its local spin density (LSD) formulation [26] reproduces well the geometries of many-particle systems, but fails in the description of their energetic features.
The introduction of the generalized gradient approximation (GGA) [27], improves considerably the reliability of the method also for the computation of the energetic parameters. In the GGA the exchange-correlation energy functional is:
where f(ρ,|∇ρ|) is a function of the density and of its gradient at a given position. Several gradient corrected exchange [28-30] and correlation [31-34] functionals have been proposed. In our study we use different combinations of those of Becke [28] and Perdew and Wang [30] for the exchange and the functionals proposed by Perdew [31] and Proynov [34] for the correlation. Recently, Becke [29] has introduced the so-called hybrid functional that is based on the “adiabatic connection” formula [35]. Because of its reliability, largely validated in the literature [36-38], we employ in our work also the B3LYP functional which uses the Becke gradient correction to the exchange [29] (B3) and the Lee-Yang-Parr [32] (LYP) one to the correlation part. The explicit formula of this potential is:
where the Ex exchange terms are that of Slater [39], that calculated by the Hartree Fock method and that proposed by Becke [29] and the Ec terms are the correlation functionals of Lee-Yang-Parr [32] and Vosko-Wilk-Nusair [40], respectively.
2.1 Reactivity Indices
The reactivity and stability of a system can be studied applying the concept of chemical hardness. Incorporation of the concepts of hardness and softness into DFT has led to the mathematical identification of η as the second derivative of the total energy with respect to the number of electrons N [41, 42]:

or, equivalently:

where the chemical potential, μ, is the first derivative of the total energy relative to the electron number. Derivatives are taken at constant external potential υ(r).
Softness is defined as the inverse of hardness:
Conventionally, the hardness is obtained from the values of the ionization potential (I) and the electron affinity (A), η = (I-A)/2, through the finite difference method. While the chemical potential is constant everywhere within the molecule, the hardness, and then the softness, is a function of the position. Thus, in addition to the global definition of η and S, the local hardness [43] and local softness [44] have been introduced.
The ab-initio SCF and DFT computations [45-52] of η are generally performed using the simple molecular orbital theory that allows one to compute the hardness as the energy difference between the highest occupied orbital (HOMO) and the lowest unoccupied orbital (LUMO) [53]:
This approximation fails if the HOMO-LUMO energy difference is close to zero. Moreover, for a more accurate computation of η, the contribution to the hardness of various orbitals should be taken into account. Recently, different approaches, accounting for the orbital hardness values, have been proposed [54-56].
The computations, presented here, are based on an approach that uses the fractional occupation number concept [57, 58]. The original idea to exploit fractional occupation numbers in the framework of DFT is due to Janak [59] who generalized the earlier work of Slater [60], using the Xα approach.
The validity of the Janak theorem in DFT, for N- and υ-representable densities, has been discussed by many authors [61-66].
In this approach, when the principle of the electronegativity equalization [67] is satisfied, the hardness matrix elements can be defined [68, 69] as the first derivative of the Kohn-Sham orbital eigenvalues (εi) with respect to the orbital occupation numbers (nj):
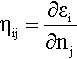
This expression takes into account the response of the i-th orbital to the change of the occupation number of the j-th orbital.
Applying the Pearson’s principle [70], that states the more localized system is less reactive, the orbital hardness can be interpreted as a measure of the degree of electron localization of a given orbital [71]. Moreover, in such a manner, the two particle interelectronic interaction, that is the most significant contribution in the hardness evaluation, is considered [69].
Numerically, the hardness matrix elements are calculated by the finite difference method [57]

The reciprocal of hardness matrix is the softness one [68] {Sij} = {ηij}− 1. Then, the absolute hardness is calculated by:

The η value is directly related to the electron localization (system stability) and the softness is associated to the reactivity indices [68, 71]. The hard (soft) systems correspond to a high (low) stability.
The formalism of DFT allows one to introduce another important local variable, the Fukui function f(r), originally defined by Parr and Yang [68] as the first derivative of the electronic chemical potential μ with respect to the external potential υ(r):

The Fukui function measures how sensitive a system’s chemical potential is to an external perturbation at a particular point. In the approach used by us, the Fukui indices are approximated by the equation:

with ![]()
Following this formalism, the chemical potential can be computed from the orbital softness values through the use of the orbital Fukui index.
2.2 Thermochemical properties
Gas-phase proton affinity (PA) is defined as the negative of the enthalpy for the process:
![]()
and can be calculated as follows:
The Ecl terms are obtained from the calculations and Evib includes the zero point energy and temperature corrections to the vibrational enthalpy derived by computed harmonic vibrational frequencies. The corrections due to translation, vibration and rotation are treated classically, using the equipartition theorem. The consideration that BH+ and B rotational contributions are quite similar, that the proton has only translational degrees of freedom and that the eventually different populations of the vibrational states that originate bringing the system from zero degrees to room temperature, are practically cancelled in the calculation of PA, yield us to consider only a further contribution of 3/2RT. Thus 5/2RT represents the translational energy of proton and the Δ(PV) = RT term necessary to convert the energy in enthalpy.
The absolute gas-phase basicity (GPB) is calculated as the negative of the standard free energy ΔG:
The entropy contribution is given by
where, at 298 K, the TS(H+) term has a value of 7.76 kcal/mol [72].
In the case of gradient corrected calculations the BH+ and B entropies have been obtained by a simple procedure that uses the theoretical harmonic frequencies and the equilibrium geometries.
Two different processes must be considered to calculate the gas-phase acidity (GPA) and the metal ion affinity (MIA), respectively:

The way to obtain the GPA is the same as in the case of GPB, whilst, the computations of MIA follows the criteria used for PA determination.
2.3 Computational details
Gaussian 94 [73], DeMon [74] and DGauss [75] packages have been employed for the calculations.
Full geometry optimizations without symmetry constraints have been executed in all cases. Vibrational analyses have been performed in the framework of internal procedures implemented in the used codes.
The five functionals employed are:
- B3LYP (Becke exchange + Lee, Yang and Parr correlation) [29, 32]
- BP (Becke exchange + Perdew correlation) [28, 31]
- PWP (Perdew and Wang exchange + Perdew correlation) [30, 31]
- BLAP3 (Becke exchange + Proynov correlation) [28, 34]
- PLAP3 (Perdew exchange + Proynov correlation) [30, 34]
The first three have been chosen because they represent the currently most popular functionals for applications to molecular systems, while the latter two because they have been used, until now, in few cases.
The basis sets selected consist of:
- The Gaussian 94 internal 6-31G** [76] to which polarization and/or diffuse functions, as specified in the Tables, have been added.
- Valence triple-ζ basis set (TZVP) of Godbout et al. [77]
- cc-pVT(Q)Z and AUG-cc-pVT(Q)T triple- and quadruple- ζ basis set of Dunning et al. [78]
For Se, Te and Ni heavy atoms the Huzinaga type-model core potentials have been used that treat explicitly the 3d10 4 s2 4p4, 5 s2 5p4 and 3p6 3d9 4 s1 electrons, respectively.
3 Results and discussion
3.1 Ion geometries
The performance of DF methods in the reproduction of reliable geometrical parameters for neutral molecules has been largely confirmed [1-10]. Usually errors of about 0.02 Å and few degrees with respect to the experimental data, are found for bond lengths and valence angles, respectively. Less data exist, in the literature, for the charged species probably because of a minor experimental information. The few density functional works [12-19] demonstrate a reasonable reliability also in this case.
We report here the structures of some cationic systems in order to contribute to the validation of DF methods.
In Figures 1 and 2, the geometrical parameters of CH2SOH+ and O3H+, obtained at PWP/TZVP, BP/TZVP and B3LYP/6-31G** levels, are reported together with other theoretical results [79, 80]. The data of protonated cytosine reported in figure 3 are obtained at BP/TZVP level and compared to the experimental ones [81, 82].



As is shown the DF gradient corrected structures are characterized by bond lengths which are sometime longer than the corresponding distances obtained by B3LYP/6-31G**, MP2/ 6-31G** [79] and CCSD/DZ + P [80] computations.
In the case of O3H+, MP2/6-31G** method is unable to distinguish between the two O-O lengths. Valence angles are, instead, of the same order in all the calculations.
For protonated cytosine (see Figure 3) it is possible to compare our BP/TZVP results with those coming from two sets of experimental determinations [81, 82]. The agreement, now, is very good. A similar concordance was found for protonated adenine [19] nucleic acid base.
3.2 Preferred attach sites
It is well known that experimental gas-phase studies are often unable to predict the preferred attach site of the proton or metal cations on a molecular system, but, this information is of fundamental importance for understanding the chemical reactivity of a given species. Thus, theoretical investigations can represent a very useful alternative to solve this problem. Reliable predictions on this matter are possible only if high level theoretical methods are employed. There are already evidences [14, 17, 19] that the DF methods are powerful tools for this type of determination.
In the schemes 1-3 three different protonation processes, that we have studied, are depicted and Table 1 collects the relative results obtained using different functionals.

Scheme 1

Scheme 2

Scheme 3
Table 1
Relative energies (in kcal/mol) at 0 K for the minima of the protonation processes depicted in the schemes 1-3.
| Species | Isomer | PWP/TZVP | B3LYP/6-311++G** | Previous works |
| O3 | a | 0.0 | 0.0 | 0.0a |
| b | 56.2 | 55.5 | 47.1a | |
| c | 58.1 | 58.7 | 58.9a | |
| S3 | a | 0.0 | 0.0 | 0.0b |
| b | 17.3 | 15.1 | 16.6b | |
| c | 33.6 | 36.3 | 35.1b | |
| Se3 | a | 0.0 | 0.0 | / |
| b | 7.6 | 11.0 | / | |
| c | 35.2 | 41.9 | / | |
| Te3 | a | 0.0 | / | / |
| b | -1.5 | / | / | |
| c | 31.5 | / | / | |
| CH2SO | a | 0.0 | 0.0 | 0.0c |
| b | 19.2 | 22.8 | 19.9c | |
| c | 48.7 | 50.2 | 52.0c | |
| C6H5NH2 | a | 2.8 | 1.9 | / |
| b | 5.8 | 4.2 | / | |
| c | 0.0 | 0.0 | / |

The protonation of X3 (X = O, S, Se, Te) species can give rise to the a, b and c isomers (see scheme 1).
Our study shows that the trans form a is favoured over the other two in the case of oxygen, sulphur and selenium but not for tellurium for which the cyclic form b is the most stable one. The energy difference between the absolute and the relative minimum b decreases significantly in going from O3 to Se3 systems and becomes negative in the Te3 one. For O3H+ and S3H+ species the comparison is possible with previous ab-initio QCISD calculations [79, 83]. The stability order of isomers is the same and the energy differences quite similar.
In the sulfine (CH2SO) molecule proton can attach, in principle, the carbon as well as the oxygen and the sulphur atoms (see Scheme 2) for obtaining three cationic-forms. Our study, performed employing the PWP gradient corrected and B3LYP hybrid functionals, shows that the structure a (in which H+ binds oxygen atom) is the most stable one followed by b (proton on carbon) and c (proton on sulphur) forms with the latter at about 50 kcal/mol above the global minimum (see Table 1).
The PWP and B3LYP results confirm the experimental hypothesis that indicates the a isomer as the most abundant and quantify the energy difference between the three stable minima.
The protonation process has been rationalized computing the orbital Fukui indices of neutral sulfine.
Following the procedure previously described and briefly reported in the method section, we found (at PWP level) that the Fukui index associated with orbital lone pair of oxygen in sulfine, has the value of 0.04 eV, while those associated with carbon and sulphur, are 0.19 and 0.21 eV, respectively. Speaking in terms of hard-soft theory of Pearson, we can indicate the orbital with the lowest Fukui index as the most reactive in the electrophilic reaction with H+.
The protonation of aniline is the object of our next example (see Scheme 3). Notwithstanding different experimental gas-phase studies [84-86] the question regarding the preferred protonation site on this molecule remains matter of controversy. The situation in solution is instead well defined. As it is shown in Table 1, both PWP and B3LYP calculations indicate that the protonation on the para-carbon is slightly favoured over that on nitrogen atom.
A recent mass-spectrometry study [86] concludes that the processes that yield c and a isomers are favoured respectively by thermodynamic and kinetics factors. Because of the small energy difference between the two minima (see Table 1), it is difficult to discriminate exactly what of the two processes is the most probable, but, the possibility that both isomers can be populated in gas-phase is reasonable.
An alternative indication can come, also in this case, from the computation of orbital Fukui indices of neutral aniline. We found a value of 0.19 and 0.06 e V for the orbitals mainly associated with nitrogen and para-carbon, respectively. So, the electrophilic attach of the proton seems to occur preferentially on the para-carbon because of its lower Fukui index value.
3.3 Proton affinity and gas-phase basicity
The determination of the proton affinity has a long history both from experimental and theoretical views. In the 1984 Lias et al. [87] compiled a comprehensive list of relative PA for a number of compounds and assigned absolute values on the basis of the best data available at that time. Subsequently the scale was considerably modified with data coming from new mass-spectrometry equilibrium and kinetic experiments [88-91].
Smith and Radom [92-94] showed that the G2(MP2) theoretical procedure is able to estimate proton affinity within a target accuracy of about 2 kcal/mol. The compounds studied by these authors are of small size (containing from 1 to 4 first row atoms) because of the relatively great computational efforts required by the method. Recently, the possibility to use the DF methods in the PA evaluation has been tested by different authors [12, 14, 17, 19] with encouraging results. From these studies it emerges that DF prediction of PA has almost the same accuracy of G2(MP2) one, but in a fraction of computer time.
In this work, the reliability of DF methods in the PA evaluation will be demonstrated firstly for small molecules and subsequently for larger systems, as amino-acids and nucleic acid bases.
Tables 2 collects the PA values for NH3 obtained employing different basis sets and exchange-correlation functionals.
Table 2
PA and GPB (in kcal/mol) at 298 K for ammonia.
| Method | PA (298 K) | ΔPA | GPB (298 K) | Ref. |
| B3LYP/6-31G** | 210.4 | 6.8 | 202.0 | this work |
| B3LYP/6-311++G** | 203.8 | 0.2 | 194.9 | this work |
| B3LYP/cc-pVTZ | 205.7 | 2.1 | 196.8 | this work |
| B3LYP/AUG-cc-pVTZ | 203.2 | -0.4 | 194.3 | this work |
| B3LYP/TZVP | 204.1 | 0.8 | 195.3 | this work |
| B3LYP/cc-pVQZ | 204.2 | 0.9 | 195.4 | this work |
| B3LYP/AUG-cc-pVQZ | 203.2 | -0.4 | 194.4 | this work |
| PWP/TZVP | 202.4 | -0.9 | 194.1 | this work |
| BP/TZVP | 202.7 | -0.6 | 194.4 | this work |
| BLAP3/TZVP | 205.7 | 2.4 | 197.4 | this work |
| PLAP3/TZVP | 209.1 | 5.8 | 200.8 | this work |
| G2 | 204.2 | 0.6 | / | 92 |
| G2(MP2) | 204.2 | 0.6 | / | 94 |
| G2(MP2,SVP) | 204.0 | 0.4 | / | 94 |
| EXP | 203.6 | 0.0 | / | 89 |

In the case of B3LYP computations the convergence of the results, as a function of the basis set dimension, has been tested using basis sets of increasing size starting from the double zeta (6-31G**) to arrive to quadruple zeta (AUG-cc-pVQZ) quality. As it is shown in Table 2, the 6-31G** basis set does not give reliable results. The introduction of the diffusion functions (6-311++G**) improves strongly the values of PA. The deviation, with respect to the experimental data, is of only 0.2 kcal/mol. From the values obtained by the larger basis sets (AUG-cc-pVTZ, AUG-cc-pVTQZ), it is evident that the convergence is reached already at 6-311++G** level. The use of TZVP valence basis set gives also results in excellent agreement with experimental evidences (the deviation is of 0.4 kcal/mol). On the basis of these results this same set TZVP has been used in connection with other exchange-correlation functionals.
PWP and BP results are close to the B3LYP one, while BLAP3 and PLAP3 computations overestimate the PA by 2.4 and 5.8 kcal/mol, respectively (the experimental value is 203.6 kcal/mol). Our best estimations well agree with high level theoretical data obtained with the G2 procedures [92].
The benchmark indicates that the PA of ammonia is very well reproduced employing the 6-311++G** or the TZVP basis sets coupled with B3LYP, PWP and BP exchange-correlation potentials. For this reason, the PA and GPB of other further compounds reported in Tables 3-5 are calculated at these levels of theory.
Table 3
PA at 298 K (in kcal/mol) for amine series.
| System | PAPwP/TZVP | PAB3LYP/6-31++G** | PAexpa | PAEXPb | PAexpc |
| NH3 | 202.4 | 203.8 | 203.3 | 204.0 | 204.8 |
| CH3NH2 | 213.0 | 214.7 | 215.3 | 214.1 | 216.1 |
| (CH3)2NH | 219.3 | 221.5 | 222.5 | 220.6 | 224.1 |
| (CH3)3 N | 226.4 | 226.7 | / | 225.1 | 229.1 |

Table 4
GPB at 298 K (in kcal/mol) and η (in eV) values for amine series.
| System | GPBPwp/TZVP | GPBB3LYP/6-311++G** | η |
| NH3 | 194.1 | 195.7 | 7.16 |
| CH3NH2 | 205.2 | 206.7 | 5.24 |
| (CH3)2NH | 211.6 | 213.5 | 4.28 |
| (CH3)3 N | 218.6 | 218.8 | 3.76 |

Table 5
PA (kcal/mol) for ozone, sulfine, glycine, alanine, cytosine, thymine, adenine and guanine.
| Species | PWP/TZVP | BP/TZVP | B3LYP/6-31 1++G** | OTHER | EXP |
| O3 | 149.4 | 146.8 | 143.4 | 149.5a | 148.0 ± 3f |
| CH2SO | 188.4 | 207.8 | 193.8 | 188.3b | 188.0g |
| Glycine | 214.2 | 213.8 | 211.5 | 213.6c | 213.6h |
| Alanine | 216.0 | 215.5 | 215.5 | 216.7d | 215.8i |
| Cytosine | / | 229.1 | / | 227.0d | 225.91 |
| Thymine | / | 208.8 | / | / | 209.01 |
| Adenine | / | 225.8 | / | / | 224.21 |
| Guanine | / | 230.3 | / | 225.8e | 227.41 |

Due to their great importance in chemistry, we have firstly considered the amine series (see Table 3).
The different experimental scales propose the same trend of basicity for these compounds but differ in the absolute values evaluation.
Both the PWP and B3LYP PA,s follow the experimental trends. In particular B3LYP results are very similar to those obtained by Lias et al. [95] and the agreement between all other data is sufficiently satisfactory.
Table 4 collects the absolute gas-phase basicity (GPB) and the global hardness (η) values obtained at PWP and B3LYP levels of theory for ammonia and some aliphatic amines.
In Figure 4 the PWP profile of GPB is reported together with the η one.

The GPB increases going from NH3 to Me3N species. Thus ammonia must have the largest proton affinity as indicated by the largest η value of 7.16 eV.
In the Figure 4 the good correlation between GPB and η behaviours is evident.
In Tables 5 and 6 we have reported our PA and GPB results for a number of compounds for which other previous theoretical [14, 80, 96, 97] and experimental [98-102] data are available.
Table 6
GPB (kcal/mol) for ozone, sulfine. glycine and alanine, and cytosine, thymine, adenine and guanine.
| Species | PWP/TZVP | BP/TZVP | B3LYP/6-311++G** | OTHER | EXP |
| O3 | 142.3 | 139.7 | 135.6 | / | / |
| CH2SO | 179.8 | 199.8 | 185.3 | / | 181.3b |
| Glycine | 206.2 | 205.8 | 203.8 | 206.7a | 206.2c |
| Alanine | 208.1 | 207.6 | 207.7 | 208.7a | 207.44d |
| Cytosine | / | 221.3 | / | / | / |
| Thymine | / | 201.3 | / | / | // |
| Adenine | / | 217.9 | / | / | / |
| Guanine | / | 222.7 | / | / | / |

For glycine and alanine amino acids, all the employed functionals and basis sets are able to reproduce correctly the experimental PA and GPB values. A very good agreement between theoretical and experimental determinations is found also in the case of the nucleic add bases at BP level. The maximum deviation from experiment is of 3.2 kcal/mol (about 1.5%) and occurs for cytosine.
For O3 and CH2SO the BP values of PA appear to be the less accurate, but the examples concerning the protonation reactions on oxygen atom and the relative PA determinations are too limited to decide about the performance of density functional methods in this case.
In Figure 5 the theoretical PA values of several nitrogen containing compounds are compared with those proposed by experimental studies.

The fitting procedure shows that, for these compounds, in which the protonation occurs on nitrogen atom, the values of PA obtained at PWP/TZVP level can be treated with confidence.
3.4 Gas-phase acidity
In Table 7 we have reported ΔHacid (the enthalpy variation for the deprotonation process) and the gas-phase acidity (GPA) at 298 K for the formic acid obtained at different levels of theory.
Table 7
GPA (in kcal/mol) at 298 K for formic acid.
| Method | ΔHacid | GPA | Ref. |
| B3LYP/6-31G** | 358.8 | 351.0 | this work |
| B3LYP/6-311++G** | 341.0 | 333.3 | this work |
| B3LYP/AUG-cc-pVTZ | 342.0 | 334.2 | this work |
| B3LYP/AUG-cc-pVQZ | 345.8 | 338.1 | this work |
| B3LYP/TZVP | 344.7 | 337.0 | this work |
| PWP/TZVP | 343.0 | 335.2 | this work |
| BP/TZVP | 345.0 | 337.3 | this work |
| BLAP3/TZVP | 348.4 | 340.7 | this work |
| PLAP3/TZVP | 346.2 | 338.5 | this work |
| GVB + Cl | 346.3 | / | 104 |
| G2 | 343.8 | / | 16 |
| G2(MP2) | 344.2 | / | 16 |
| G2(MP2,SVP) | 344.1 | / | 16 |
| EXP | 345 ± 2 | / | 103 |

The use of B3LYP functional with the 6-31G** basis set is, as it is evident, insufficient to give reliable results of acidity: the error with respect to experimental value [103] is of 13.8 kcal/mol. A significant reduction of the discrepancy is obtained, gradually, improving the basis sets: the B3LYP/6-311++G**, /AUG-cc-pVTZ and /AUG-cc-pVQZ computations are affected by errors of about 4.0, 3.0 and 0.8 kcal/mol, respectively.
If the TZVP basis set is used in connection with hybrid potentials, the ΔHacid value becomes 344.7 kcal/mol. The BP/TZVP and PWP/TZVP gradient corrected computations give ΔHacid of 345.0 and 343.0 kcal/mol, respectively. BLAP3/TZVP and PLAP3/TZVP ΔHacid deviate from experimental values by 3.4 and 1.2 kcal/mol. Considering that the experimental uncertainty is of ± 2 kcal/mol, almost all our results seem to be reliable as well as those obtained with GVB [104] and G2 [16] procedures. A good convergence is reached already with the TZVP basis set. The importance of this result lies in the possibility to compute GPA for larger system with acceptable computer efforts.
The calculations performed for acetic and propanoic acids (see Table 8) confirm the opportunity to use the medium-sized TZVP basis set to obtain good results. On the other hand the lack of other theoretical data regarding the propanoic acid reflects the difficulty to treat, with expensive procedures, such a kind of system.
Table 8
GPA at 298 K (in kcal/mol) for acetic and propanoic acids.
| ΔHacid | GPA | ΔHacid | GPA | |
| Method | CH3COOH | CH3CH2COOH | ||
| B 3 L YP/6-311++G * * | 345.0 | 337.2 | 345.2 | 337.5 |
| B3LYP/TZVP | 347.5 | 339.7 | 347.5 | 339.7 |
| PWP/TZVP | 346.5 | 338.7 | 345.9 | 338.1 |
| BP/TZVP | 348.5 | 340.8 | 350.9 | 343.2 |
| GVB + CI | 352.1a | / | / | / |
| MP2 | 347.0b | / | / | / |
| G2(MP2) | 346. lb | / | / | / |
| EXP | 348. ± 3c | / | / | 340.3 ± 2c |

The relationship between total hardness and gas-phase acidity has been investigated in the case of the aliphatic alcohol series. Data are reported in Table 9 and depicted in Figure 6.
Table 9
GPA at 298 K (in kcal/mol) and global hardness (in eV) values for alcohol series.
| System | GPAB3LYP/6-3I1++G** | GPAexp | η |
| MeOH | 371.6 | 375.0a | 7.60 |
| EtOH | 368.3 | 370.7a | 6.25 |
| n-PrOH | 367.8 | 368.lb | 5.67 |
| n-BuOH | 367.4 | 367. lb | 5.11 |
| n-PeOH | 367.1 | 366.3b | 4.48 |


It is possible to note that:
- theoretical and experimental values are in good agreement;
- the absolute GPA values decrease in going from MeOH to n-PeOH, consequently the acidity of the ROH increases;
- global hardness decreases underlying a lower affinity for the proton in the series.
Finally we have computed the GPA for glycine and alanine amino-acids. The results are collected in Table 10 together with the available experimental data. Single point calculations on the optimized MP2/6-31G* geometry have been performed at MP2 and MP4 levels with different basis sets (see Table 10).
Table 10
GPA (in kcal/mol) at 298 K for glycine and alanine amino-acids.
| Method | ΔHacid | GPA | ΔHacid | GPA |
| Glycine | Alanine | |||
| MP2/6-311 + G(3df,2p) | 340.2 | 332.4 | 339.6 | 331.8a |
| MP4/6-311 + G** | 344.2 | 336.4 | .343.7 | 335.9a |
| B3LYP/6-311 + G** | 340.8 | 333.0 | 340.3 | 332.5 |
| B3LYP/6-311++G** | 338.7 | 331.9 | 3.39.1 | 331.6 |
| PWP-TZVP | 339.8 | 332.1 | 339.0 | 331.2 |
| BP-TZVP | 345.3 | 337.5 | 342.3 | 334.6 |
| G2 | 342.1 | 334.3 | 341.5 | 3.33.8a |
| EXP | 342.3 | 335.4 | 341.5 | 334.5b |

Taking as reference the measured value [106| of 342.3 kcal/mol for glycine and 341.5 kcal/mol for alanine, and considering that experimental ΔG were obtained by assuming a fixed TΔS (6.9 kcal/mol) that is lower than all other calculated values for both molecules, all our theoretical values can be defined quite good. In particular the G2 procedure gives a difference of -1.1 kcal/mol between the experimental and theoretical values in the case of glycine and -0.7 kcal/mol for alanine [107].
MP4/6-311 + G** computations give errors of 1.0 (glycine) and 1.4 (alanine) kcal/mol.
At the MP2/6-311 + G(d,p) level, ΔG deviations of -3.0 and -2.7 kcal/mol for glycine and alanine are found, respectively.
The deviations resulting from the B3LYP/6-311 + G** (6-311++G**) computations are: -2.4 (-3.5) kcal/mol for glycine and -2.0 (-2.9) kcal/mol for alanine.
The gradient-corrected PWP/TZVP functional gives differences of -3.3 kcal/mol for both glycine and alanine. Finally the BP/TZVP ΔG values deviate from the experimental ones by 2.1 kcal/mol for glycine 0.1 kcal/mol for alanine.
3.5 Metal ion affinity
The determination of the metal ions affinity for the organic bases is important to elucidate the acid-base reaction mechanism as well as to obtain information about fundamental biochemical processes (i.e. synthesis, replication structural integrity and cleavage of DNA and RNA) [108].
Because of the presence of the metal, theoretical investigation is often difficult and requires an appropriate treatment of electron correlation and the use of opportune basis sets.
Recently Del Bene [109] has investigated the basis set and the correlation energy effect on lithium ion affinities for a series of first-and second-row neutral bases employing the Hartree-Fock and the Moller-Plesset perturbation methods.
We have performed similar calculations, in the framework of density functional theory, with the aim to define the basis set convergence limit and the influence of the exchange-correlation potential on the interaction of lithium cation with ammonia.
Data are reported in Table 11 together with previous theoretical [109] and experimental [110] results.
Table 11
MIA at 298 K (in kcal/mol) of Li+ for NH3
| Method | MIA | Ref. |
| B3LYP/6-31G** | 45.6 | this work |
| B3LYP/6-311++G** | 40.1 | this work |
| B3LYP/cc-pVTZ | 41.4 | this work |
| B3LYP/AUG-cc-pVTZ | 39.1 | this work |
| B3LYP/TZVP | 37.4 | this work |
| B3LYP/cc-pVQZ | 40.1 | this work |
| B3LYP/AUG-cc-pVQZ | 39.3 | this work |
| PWP/TZVP | 36.6 | this work |
| BP/TZVP | 28.3 | this work |
| BLAP3/TZVP | 29.9 | this work |
| PLAP3/TZVP | 36.5 | this work |
| MP2/cc-pVDZ | 45.8 | 109 |
| MP2/cc-pVTZ | 41.2 | 109 |
| MP2/cc-pVQZ | 39.8 | 109 |
| MP2/AUG'-cc-pVDZ | 38.6 | 109 |
| MP2/AUG'-cc-pVTZ | 38.7 | 109 |
| MP2/AUG'-cc-pVQZ | 38.9 | 109 |
| MP4/cc-pVDZ | 45.4 | 109 |
| MP4/cc-pVTZ | 41.1 | 109 |
| MP4/AUG'-cc-pVDZ | 38.4 | 109 |
| MP4/AUG'-cc-pVTZ(-df) | 39.1 | 109 |
| MP4/AUG'-cc-pVTZ | 38.6 | 109 |
| HF/AUG'-cc-pVTZ | 39.8 | 109 |
| CCSD(T)/AUG'-cc-pVTZ | 38.7 | 109 |
| EXP | 39.1 | 110 |
The B3LYP computations are strongly influenced by the basis set quality. In fact, as in other cases, the 6-31G** set does not describe well the interaction. Our data, together with those reported in literature and included in Table 11, provide further evidences about the importance of the inclusion of diffuse functions on non-hydrogen atoms in order to obtain better agreement with experiment. From the Table it is clear that, good accuracy, can be reached employing medium sized basis sets. Using the same TZVP bases, we have tested the role of the exchange-correlation functionals in the determination of Li+ affinity for NH3. Comparison with measured value reveals that B3LYP/TZVP gives the better MIA (37.4 vs 39.1 kcal/mol) followed by PWP/TZVP (36.6 kcal/mol) and BP/TZVP (28.3 kcal/mol). Both the calculations performed by the LAP3 functional do not differ significantly with respect to those executed at PWP and BP levels, respectively. In other words, this fact, could mean that the choice of the exchange functional is important.
On the basis of the performed benchmark on ammonia, we have extended the study to the first terms of aliphatic amine series, using only the B3LYP/6-311++G** computational procedure. The results are collected in Table 12. The theoretical values follow strictly the experimental trend.
Table 12
Absolute Li+ affinities (MIA) at 298 K (in kcal/mol).
| System | MIAB3LYP/6-31++G** | MIAexpa |
| NH3 | 40.1 | 39.1 |
| CH3NH2 | 41.5 | 41.1 |
| (CH3)2NH | 42.3 | 42.2 |
| (CH3)3 N | 41.7 | 42.1 |
Taking into account that experimental absolute lithium affinities are accurate to ± 2 kcal/mol [110], and that the values referred to the different amines tire very close, we can attribute to the used method a good reliability in the reproduction of these parameters.
The next example regards the determination of Li+ and Na+ ions affinity for glycine and cytosine. Because our aim is also that to establish what functional can have general reliability for all kind of systems, we have used, in this case, the PWP/TZVP combination. On the other hand this choice is easily justified by the similar MIA values of Table 11.
Table 13 resumes the results obtained. We can note that for glycine our values are quite similar to the measured ones. In the case of cytosine the MIA,s are overestimated by about 10 kcal/mol. This fact can be explained considering that Li+ and Na+ cations form a bridge bond with the N1 and O2 atoms of the ring, determining a redistribution of the net charges and consequently a bond order variation that, neither PWP/TZVP nor B3LYP/6-311++G** calculations are able to take into account.
Table 13
Li+ and Na+ MIA (kcal/mol) at 298 K for glycine and cytosine.
| System | MIApwp/tzvp | MIAexp |
| Gly-Li+ | 54.2 | 51.0a, 51.91b |
| Gly-Na+ | 42.1 | 39.4a, 38.0b |
| Cyt-Li+ | 62.5 | 55.5a |
| Cyt-Na+ | 53.2 | 42.3a |
Finally we have performed some calculations in order to verify the performance of density functional methods in the prediction of MIA when transition metals are involved. Because of the availability of the experimental information [113], we have chosen to investigate the interaction of Ni+ cation with ammonia, methyl- and ethyl-ammonia. The relative metal ion affinity (ΔMIA) values tire shown in the Scheme 4 and compared with experimental counterparts. The dissociation limit for Ni+-amine systems is represented by the metal ion in the its ground state (2D) and the free amines. As it is evident B3LYP/6-311++G** computation reproduces only the trend proposed by the experiment, but the absolute values are strongly overestimated. The same behaviour has been obtained with the PWP functional. The overestimation is related to the well known fact that density functional methods fail in the description of electronic states of transition metal isolated atoms [7-9]. For this reason, present results must be considered as qualitative rather than quantitative indications.

Scheme 4
3.6 Potential energy surfaces
The study of reaction paths, in DFT, is not a new [114, 115]. Thus we have chosen to explore the potential energy surfaces (PES) introducing the possibility to rationalize the results through the computations of the global hardnesses along the whole reaction path, with the aim to verify if, for the studied processes, the maximum hardness principle (MHP) [53] is satisfied.
In the scheme 5 are depicted the stationary points of O3 and S3 protonation paths studied at PWP/TZVP level.

Scheme 5
Energetic and hardness profiles are drawn in Figures 7 and 8, respectively.


From the energetic point of view the trails isomer 2 is the global minimum in the PES of both processes followed by 1 and 3 structures. Two transition states connect the 1-2 (TS1) and 2-3 (TS2) isomers.
Following the MHP principle, we must expect that the energetic and hardness curves, as a function of the reaction coordinate, be specular, if the chemical potential does not change significantly along the reaction paths. As is evident from Figure 7 and 8, this circumstance is verified for both the studied processes.
In particular the maximum hardness value is found for the absolute minimum and decreases in correspondence of relative minima and transition states following the stability order.
4 Conclusions
From the results presented above, we make the following conclusions:
- In the determination of ions geometries, the comparison with experimental data, underlines, in some case, the importance of the employed functional in determining reliable structures.
- Accurate information on preferred attack sites and potential energy surfaces can be obtained with computational costs lower than those required by HF methods including configuration interaction.
- Thermochemical properties are well reproduced even with medium-sized basis sets such as the 6-311++G** and TZVP.
- Metal ions affinities, for transition metal containing systems, can be defined good from a qualitative point of view, but, the absolute values are not so accurate and require improvements in the computation of atomic energy.
- DFT based descriptors of reactivity are powerful tools for the explanation of all examined processes.
Acknowledgements
We are grateful to the Italian MURST and CNR and Bulgarian National Scientific Foundation for financial support.