
Half a Century Of Hybridization‡
C. Barbier*; G. Berthier** (*) U.F.R. de Chimie-Biochimie, Université Claude Bernard Lyon 1 43 Boulevard du 11 Novembre 1918, F 69622 Villeurbanne cedex
(**) Laboratoire d’Etude Théorique des Milieux Extrêmes Ecole Normale Supérieure, 24 rue Lhomond, F 75231 Paris cedex 05
Summary
The hybridization principle of atomic orbitals used from the thirties up to the present days as a prelude to the formation of chemical bonds is surveyed. The expose is centered on the ab-initio procedure of overlap-matrix localization suggested by G. Del Re in 1963 (Theoretica Chimica Acta 1, pp 188-197), and its successive extensions and various applications in Quantum Chemistry are described. Some conceptual aspects of hybridization are discussed.
1 Introduction : A perennial concept
Through its ups and downs, the concept of orbital hybridization is most significative for the perspectives opened out by Quantum Chemistry from about 1930. Let us only mention all the disputes in which it was involved during almost one century with regard to tricky questions of molecular structure (i.e., the supremacy of valence-bond pictures over molecular orbitals or vice-versa, the physical content of the resonance theory, the localization versus delocalization dilemma and so on…). Furthermore, its study gives us a good example of the specificity of the scientific explanation among chemists (i.e., the rejection of reductionism to Physics [1]).
1.1 The first period
The algebraic manipulation of atomic orbitals to which the name of hybridization has been attached was initiated by Pauling [2]. Merging together the idea of shared electron pairs of the classical valence theory of Lewis and the quantum mechanical treatment newly presented by Heitler and London for the hydrogen molecule [3], he offered a somewhat intuitive description of the chemical bond in complex molecules by using “hybrid orbitals of maximum bond-forming power” [4]. More concretely, the four valences of the carbon atom in the methane molecule were assigned to four sp3 orbitals whose axes of maximum electron density were arranged in conformity with the dictum [5] “There is a large amount of experimental evidence suggesting that the carbon atom has four valencies radiating from the centre to the corners of a regular tetrahedron”.
In subsequent independent papers, Pauling [4] and Slater [6] generalized the valence-bond treatment made for the H2 molecule to polyatomic systems as H2O, NH3, CH4 etc … where an atom of the first period (the second row) is linked to hydrogens by several two-electron bonds ; they described the valence orbitals coming from the central atom by appropriate s and p combinations known later as hybrid orbitals. At the same time Hund [7] and Mulliken [8] presented another quantum theory of valence, the molecular orbital method in LCAO form, using the spectroscopic concept of molecular configuration built from s, p, d …pure atomic orbitals. The actual status of the hybridization process was clarified by Van Vleck [9], who showed that the various approximations introduced in the quantum mechanical treatment of valence were responsible for many differences of interpretation rather than the content of the methods themselves. For instance, hybridization involved in the perfect pairing of localized electron-pairs and resonance limited to chemically significant bond functions amount the same thing [10,11] This culminates in the final equivalence of methods starting from common sets of -pure or mixed- atomic orbitals (i.e., valence bond or molecular orbital treatments including all the possible distributions of electrons in the wave function) first proved for very simple systems [12,13] and later generalized [14,15].
1.2 Valence state theory
From the point of view of Natural Philosophy, the concept of atomic valence states, developed by Van Vleck, Mulliken and others [9,16,17,18] in close connection with hybridization, is a major achievement, for it gives a microscopic picture of the atom in situ taking into account the level of complexity to be preserved in the theoretical analysis of chemical facts [1]. The basic ingredient of the theory consists in a formal decoupling of some paired electrons for the atom considered in accordance with its valence in molecules : To do that, two steps may be necessary : first, promoting one electron of a 2s filled subshell, if necessary, into the adjacent p shell (case of carbon s2p2 versus s1p1p1) or d shell (case of transition metals s2dn−2 versus s1dn−1) ; second, randomizing the spins of the valence electrons with respect to each other (i.e., by means of the value 1/2 Kij assigned to the exchange term corresponding to the valence orbitals of the atoms in question [16]).
The valence state definition above is just operative, for it generates energy terms calculable from spectroscopic data in the frame of the Slater model of atoms in their neutral and adjacent ionized states. The valence orbitals can be defined in pure or hybrid forms, corresponding to familiar s,p tetrahedral (te), trigonal (tr) and digonal (di) sets for light atoms, or to the huge variety of s,p,d combinations for heavier atoms [19,20]. To conclude by a fairly common remark on the valence states, we will add the fact that their building-up process from atomic experimental data, the Fk and Gk Slater parameters [21], implies that they do not be thought as physical observables, but rather to a linear combination of genuine spectroscopic states [22]. Without going into details of chemistry, the energy loss due to the contribution of atomic excited states in this mixture is said to be largely paid by the formation of bonds.
1.3 The present status of hybridization
Nowadays, the concept of hybridization remains the basis of the most popular description of the molecular bonding, namely that of σ, π - and δ -chemical bonds between atoms in organic [23] as well as inorganic compounds and solids [24]. The terminology of hybridization gives us a simple way to characterize “atoms in molecules” [25] by their valence states, which in the same time precises the type of geometry of the atomic site considered. We can use hybridization not only to sketch the spatial distribution of the binding electron pairs, but also according to Pauling to describe lone pairs with privileged directions depending on their percentage of s and p characters [26,27]. If, finally, we pass over the questions connected to its theoretical origin, hybridization can be considered as a simple way for describing, both in speaking and in writing, the so-called molecular observables in terms of atomic components.
Another reason for the success of such interpretations lies in the fact that hybridization becomes a flexible model applicable to systems of arbitrary shape when it is equipped with the maximum overlapping principle establishing a link between bond strengths and overlap integrals of hybrid orbitals centered on neighbouring atoms [28,29]. As it will be shown in the next section, this is the guiding idea of Murrell [30], Coulson [31] and Del Re [32] for a priori calculations of hybrids in a molecule. In the seventies, however, hybridization has been fiercely criticized by people pretexting that its use supposes a preliminary knowledge of the molecular structure to be predicted and preferring the VSEPR orbital-free model of Gillespie instead [33]. In so far as a qualitative picture of the molecular structure is requested, it would be better to say that both models are basically isomorphous, mimicking molecules by a set of initially equivalent sites whose interaction is afterwards governed by an energy criterion (i.e., the maximum overlapping principle of hybridization, or the hierarchy of electron-pair repulsions in the VSEPR model [34]). By no means, the quantum-chemical applications of the hybridization concept are really concerned in this quarrel ignoring the fact it is first of all a procedure intended to prepare atoms in a molecule to bonding. To end the matter, suffice it to speak in measured terms of hybridization : i) it is a way for constructing “chemical orbitals” , that is to say wave functions which preserve the concept of bond properties [35,36] and in the same time gives us good starting points for the developpement of various semi-empirical or ab initio treatments-, ii) it is not an experimentally observable phenomenon, but according to Coulson merely “a feature of a theoretical description”.
2 Theoretical determination methods of hybrid orbitals
2.1 Geometrical constructions
To avoid possible problems of linear dependence, it is generally assumed that the hybrid orbitals hAi centered on a given atomic site A are orthonormal:
If so, the direction and form of hybrids starting from the central atom A of a highly symmetrical system, as methane or complexes of transition metals, for instance [Co(NH3)6]3 −, are defined by the geometry of the molecule, independently of the nature of the nearest neighbours B. In such cases, hybridization of orbitals on atom A gives us a convenient basis for describing the set of equivalent bonds A-B by combination with the orbitals of atoms B ; hybrids of A adapted to the symmetry of the molecule (i.e., to its directed valences) are easily determined by projecting a set of central-field orbitals s,p,d … of atom A into the irreducible representations of the point group to which the molecule belongs [19]. Lists of all the possible types of s,p,d hybrids for highly symmetrical molecules and a number of s, p, d, f combinations are given in the literature [37,38]. Hybrids for some compounds of lower symmetry have been also reported [39,40].
More generally, hybrids pointing in any direction around an atomic site A can be obtained by straightforward geometrical constructions [41] provided the form of the molecule is already known (i.e., by another theoretical or experimental way). The vectorial character of the px, py, pz orbitals transforming as the axes of a cartesian coordinate system ensures the success of such calculations, except if the valence angles become smaller than 90° : A well known example is cyclopropane, a three-membered ring with bond angles of 60° to be described according to Coulson and Moffitt [42] by a model of hybridized “banana bonds” explaining its conjugation properties as a substituent on aromatic nuclei. A somewhat similar situation occurs with four-membered rings, as cyclobutane [43]. It has been suggested [44] that the difficulties leading to bent banana bonds in small rings could be circumvented by accepting combinations with complex coefficients as solutions for hybrids. The difficulties due to the occurrence of broken symmetry solutions with respect to time reversibility when using complex wave functions can be solved by separating real and imaginary parts in variational caculations [45]. However, bent bonds formed from real orbitals are a little more convenient [46].
If we limit ourselves to s and p combinations, we can state without any doubt that the bonds formed between an atom A and its neighbours B using appropriate geometrical hybrids verify the principle of “maximum overlapping of atomic orbitals” (M.O.A.O. criterion) even if the atoms B linked to A are different. Clearly, the integrated product hAhB of a pair of σ hybrid orbitals giving the best bond energy EAB will be as large as possible if the directions of hA and Jig in space coincide, the overlap between hA and hB being maximum. However, hybridization of d (and f ) put some problems to theoreticians, for instance the preservation of the cylindrical symmetry properties of s p d hybrids [47] and the unicity of possible solutions [48], but they are not serious according to Pauling [49]. The alternative suggested by Daudel and Bucher [50] was relaxing the orthogonality constraints.
2.2 Maximum overlap criteria : The Del Re method
Methods for a priori determination of atomic hybrids using the principle of maximum overlapping have been implemented towards the sixties, either in global form or in a more local form. The procedures used by Murrell for quasi centro-symmetrical molecules [30] and by Coulson and Goodwin [31] consist in simply maximizing the sum of all overlap integrals between σ paired orbitals. More or less different techniques have been suggested later [51,52] The method developed by Del Re from 1963 [32] is a more refined multi-local treatment involving each atom and its next neighbours in turn. Taking the likeness of the eigenvectors of the overlap and effective Hamiltonian matrices for granted as a consequence of the possibility of evaluating off-diagonal terms approximately by means of the Mulliken proportionality rule to overlap (see [53,54]), this procedure is based on a principle of “maximum localization of hybrid orbitals” (M.L.H.O. criterion). The best hybrids are the ones which lead to a localization of the overlap matrix of the whole molecule, under the condition that the set of orbitals assigned in this manner to each atom be orthonormal. Here, the word “localization” means that the resulting Hamiltonian is -to a certain degree of approximation- factorized in 2×2 blocks corresponding to the various pairs of bonded atoms, except if the presence of several large off-diagonal elements suggests the idea of many-center bonds. Such a definition gives us a unique set of hybridized functions for each atom of a molecule consistent with the maximum overlapping criterion and in the same time an efficient computational procedure for any system. The equations of the Del Re hybridization method have been written out first in the case of single-zeta s, p valence orbitals of first-row atoms and generalized later to more complicated basis sets, as it will be discussed below.
2.2.1 Building procedure of maximum localization hybrid orbitals
Consider a system composed of atoms A, B, C etc … and let χA, χB, χC be their respective 2s, 2px, 2py , 2pz valence orbitals defined in a single coordinate system. The basis set of the whole molecule can be represented by the row vector χ = (χA , χB , χC etc …) collecting all these basis functions :
The overlap matrix S = (χ†, χ) formed of diagonal elements AA, BB all equal to the 4×4 unit matrix I and of off-diagonal blocks AB, AC etc … such as SAB = (χ†A, χΒ) can be written as follows :

where SAB, SACetc … are the overlap matrices between pure atomic orbitals of A , B , C etc …
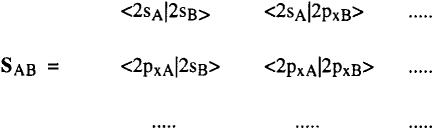
We have to find a unitary transformation U changing the χ basis into a new one χ’ such as U only mixes the orbitals belonging to a same atom without mixing them with those of other atoms. Then the χ’ basis is changed into
and SAB and S into

The localization would be perfect if each of the off-diagonal blocks S′AB, S′AC… contains only one non-zero element on its diagonal, say λAB λAC… , for a given hybrid of atom A should form a bond with only one hybrid belonging to each of its neighbouring atoms B, C … Therefore, the U transformation has to keep the diagonal blocks of S unchanged and to transform the remaining part of the S matrix in order to have only one element in each row and column. It is always possible to arrange the hybrid orbitals of any (polyatomic) molecule in such a way that the overlaps < hAB | hBA > , < hAC | hCA > etc … are on the diagonal of their S′AB S′AC etc… respective blocks. The maximum overlap condition will be practically satisfied if all the S′xy blocks are as close as possible to diagonal matrices, that is to say if the S′ whole matrix has the following form:

where the terms of the off-diagonal blocks explicitely written are those having the largest absolute values for bond-hybrid overlaps. For instance, we have:
![]()
with λBA = λAB. Putting U†A1 and U†Bl for the first rows of U†A and U†B , that is to say :

and multiplying Eqs (10) and (11) on the right by U†B and U†A respectively, we get
or after multiplying Eq. (12a) by S†AB, substituting Eq. (12b) and transposing :
Similarly,
![]()
Thus, UA1 and UB1 are the eigenvectors associated with the largest eigenvalues of SABS†AB and S†ABSAB respectively. If we now pass to the pair of atoms A C, we can apply the same treatment to the S′AC block matrices, so that we have a new pair of equations defining UA2 and UC1 and so on … This process has be repeated for each atom, giving a number of conditions depending on its number of valence orbitals and next-neighbours (here, four).
The eigenvectors computed from the largest eigenvalues of Eq. (13), say ÛA1, ÛA2, ÛA3, ÛA4, ÛB1, ÛB2, ÛB3, ÛB4 etc …, give only a first approximation Û of the unitary transformation U we wanted. It is due to the fact that the local transformations ÛA, ÛB defined in this way for the various atoms A, B etc…are not unitary because the overlap matrices connecting each of them with its neighbours do not generally commute. This deficiency can be cured by substituting unitary transformations UA, UB for ÛA, ÛB etc … as close as possible from the latter (i.e., by requiring that the scalar product of the ÛA and UA vectors is maximum) Using Eq. (13), the corresponding variational problem writes:
with a symmetrical set MA of Lagrange multipliers Mij preserving the unitary character of the UA transformations. Differentiating Eq. (14), we get a couple of transposed equations such as:
where ![]() is a diagonal matrix of elements
is a diagonal matrix of elements
We can solve them by assuming that the UA blocks forming the U unknown transformation are the products of two new unitary matrices VA and W†A , the first one permitting by the same token to reduce MA to its diagonal form mA:
Then, Eq. (15) and its transpose are replaced by
equivalent to
Multiplying Eq. (18b) by Eq. (19a) and Eq. (18a) by Eq. (19b) on each side, we obtain two eigenvalue equations, namely :
the resolution of which gives WA and VA , that is to say the hybridization matrix UA = VA W†A for each atom A we were looking for. However, it is necessary to make sure that the elements of the mA matrix as calculated from Eq. (19a) have non-negative values, a condition which may be always ensured by changing signs in the columns of WA initially computed from Eq. (19b) [32].
In Fig.1, we give the flow chart of the computer program to be implemented in order to determine the best possible hybridization matrix U for a molecule satisfying the criterion of maximum overlap according to Del Re. The successive steps of the calculation for a given atom, say A , are :

1. Construct the overlap matrices SAB, SAC …
2. Compute SAB S†AB and S†ABSAB, SAC S†AC and S†AC SAC etc …
3. Evaluate the eigenvalues λ2 of the couples of AB , AC etc … product matrices
4. Select the highest eigenvalues (λ2AB)max, (λ2AC)max,… and the corresponding eigenvectors and form the ÛA blocks, the columns of which are the eigenvectors associated with (λ2AB)max, (λ2AC)max etc …
5. Form the diagonal matrix Λ from the λ squared precedent values
6. Calculate and diagonalize the Λ2a Û†A ÛA Λ2A product , the eigenvectors of which are the columns of the new matrix WA and the eigenvalues are the elements mAi2.
7. Calculate and diagonalize the ÛA Λ4A ÛA product, the eigenvectors of which are the columns of the new matrix VA.
8. Form the product VA W†A = UA giving the hybridization matrix of the orbital centered on atom A (and the angles between resulting hybrids from their s and p percentages)
9. Repeat for the next atom B and so on …
The algorithm of Fig. 1 works satisfactorily as soon as atom A is able to use all its valence orbitals to form as many bond hybrids with its partners B, because a well-defined non-zero λAi2 value is effectively found for each hybrid. In the case of an insufficient number of partners B as in Fig.1, the zero input-values assigned to orthogonal orbitals corresponding to absent links have to be treated in different ways according to the structure of the molecule considered : i) A belongs to a conjugated system ; λ2Ai= 0 corresponds to a (2)pπ pure atomic orbital ii) A is a heteroatom bearing a σ lone-pair, as nitrogen in amines or oxygen in carbonyls; λ2Ai = 0 has to be substituted by an appropriate non-zero value in order to have a set of hybrids correctly directed [55], as will be described in Section 2.2.2 iii) A is a radicalic center, as in trivalent carbon compound ; λ2Ai = 0 is retained or substituted by another value, according as the species in question is a π or a σ free radical.
In passing, we will note that the algorithm first developed for hybridization can be adapted to the problem of the localization of the normal vibration modes given by the GF Wilson method [56].
2.2.2 The lone-pair problem
The sole maximum localization criterion is unable to give physically significant results for the σ lone pair of a heteroatom because the not-overlapping hybrid corresponding to it is completely determined by the orthogonalization condition with the bond hybrids. As a result, the 2s valence orbital is ill-balanced between the components of the whole set of resulting hybridized functions, and so the angles between them is in poor agreement with the experimental bond angles ; for instance, the bonding hybrids of water appear to have a high s-character, so that they form a too large angle (138° versus 105° for the experimental bond angle). In such cases the maximum localization criterion strictly limited to bond overlaps (i.e., without any restriction on nonbonding hybrids, except for the orthogonalisation condition) yields undesirable bent bonds.
The situation is drastically changed by introducing new conditions on non-bonding electrons, for instance on energetical reasons. According to considerations going back to Pauling, the fact that non-shared electrons of an atom have the propensity of occupying lower energy levels and, consequently, hybrid orbitals whose s-character is more important that of the bonding ones, can be envisaged. This was, in fact, the origin of the hierarchy of the electron-pair repulsion terms in the Gillespie-Nyholm model [33].
The solution retained by Del Re et al. was to treat the question in a heuristic suitable way. The non-bonding orbitals of A are determined as if they were overlapping with a non-specified orbital belonging to a phantom partner B’ in addition to its real neighbours B1 , B2, … The corresponding λ2Anb value between, say 0 and 0.5 , was adjusted by plotting the angle of the resulting hybrids, to be associated to the bonds between A and B1 , B2 , … versus the experimental angle B1AB2 in reference molecules. Using ordinary Slater orbitals for the overlap integrals requested by the Del Re method, it is found that the value λ2nb = 0.25 give bond hybrids practically equal to the experimental bond angles in water and ammonia, whereas they are slightly smaller than their experimental counterparts in some methyl derivatives.
In computer programs using hybridization routines, an automatic procedure for evaluating the weights λ2Anb assigned to the σ lone-pair hybrids of atom A has been devised, namely the arithmetic mean of the weights λ2AB corresponding to the set of bond hybrids from A divided by a scale factor κ. Different values have been suggested for κ, 1.45 [57] or 1.30 [58] according to the method in which hybridization has been implemented. For generality’s sake, λ2Anb is finally multiplied by the number of electron pairs occupying the nonbonding hybrids : 0 for a vacant hybrid, 0.5 for a σ hybrid with a single non-paired electron (σ free radicals), 1.0 for a doubly occupied hybrid (σ lone pairs).
A non-empirical alternative supporting a value λ2Anb close to 0.2 which has been adopted for lone-pair weights in the preceding treatment was presented later [59], using the concept of eccentric functions to enlarge the valence basis set of heteroatoms by means of lone-pair functions (LPF). The NH3 molecule was studied as a test without and with a LPF in the vicinity of nitrogen. A reference calculation with Slater orbitals of exponents ζ2sN = ζ2pN = 1.90 , ζ1s= 1.24 for valence orbitals gives bond hybrids reproducing the experimental bond angle HNH provided a weight λ2Nnb= 0.21 is assigned to the lone-pair hybrid. If a 1s hydrogenic function of exponent ζLP is located on the C3 symmetry axis at a non-zero distance RLP from the nitrogen atom, the standard hybridization procedure gives a complete set of non-zero eigenvalues λ2Ai , hence the possibility of having correct bond hybrids by adjusting the parameters ζLP and RLP properly. The best result, obtained with a semi-diffuse LPF (ζLP = 0.3) close to nitrogen (RLP = 0.1 a.u.), compares fairly well with the primitive empirical procedure.
2.2.3 Extensions to enlarged basis sets
The hybridization procedure described in Section 2.2.1 is fulfilled by a sequence of block transformations starting with overlap matrices SAB generated, for convenience, by minimal sets of s and p orbitals. This particularity, however, does not create serious difficulties for possible extensions to more general basis sets. The 4×4 dimensions of the block matrices originally considered have to be only increased in conformity with the size of the bases in question.
The addition of five d orbitals to an s and p set, either as extra orbitals in the second period (the third row) from silicium to chlorine, or as valence orbitals for transition metal complexes is taking into account straightforwardly by raising the dimensions of the block matrices up to nine [60]. However, mixing the d functions added to the valence orbitals of non-metals, for instance sulphur, or keeping them in their primitive form, has not a great importance, because they play just a role of polarization functions in molecular structure calculations using hybridization [61].
The case of atoms described by extended basis sets with several s and p functions per shell is a little more complicated [62]. For instance, let us consider the split-valence sets built in double-zeta form from Slater or contracted Gaussian orbitals, which are commonly used for ab-initio calculations of organic compounds. An example is the very popular 3-21 G and 6-31 G sets of Pople implemented in current computer programs of Quantum Chemistry. For each atom, this description involves an inner group of rather concentrated functions 2s and 2p and an outer group of more diffuse functions 2s’ and 2p’ (large exponents and small exponents, respectively). First of all, it is necessary to begin the procedure by orthogonalizing the inner and outer functions 2s and 2s’, 2p and 2p’ centered on a same atom A, for the generally important one-center overlap between the inner and the outer sets should destroy the meaning of any hybridization method based on the values of two-center overlap integrals. The choice of a definite orthogonalization transformation for this purpose, e.g. those of Schmidt or Löwdin, is immaterial. It is then possible to perform all the transformations sketched in Section 2.2.2 after doubling the 4×4 size of the various block-matrices indicated on the chart of Fig. 1. The eigenvectors corresponding to the lowest eigenvalue λ2i give hybridized double-zeta orbitals, to be assigned to bonds and lone pairs as usual, whereas the existence of a second set of hybridized orbitals corresponding to the next eigenvalue λ2i suggests other possible applications, for example in genuine double-zeta valence bond calculations.
Other hybridization procedures for double-zeta basis sets may be imagined, for instance by using projection techniques [63] : Starting with a set of hybrids obtained from a parent minimal basis (e.g., STO 3G versus 6-31 G) we can project the latter hybrids into the space spanned either by the inner subset or the outer subset of the original split-valence basis. The extension of the Del Re algorithm for hybridization to multiple-zeta sets does not raise any major difficulty, which illustrates both the flexibility of the method and the perenniality of the concept.
3 Conclusions : A multiple-purpose instrument
3.1 Miscellaneous structural applications
For a long time, and still nowadays, hybridization has been used as a starting point for, so to say, a “linnean classification” of molecular observables ; it enables us to rationalize a large body of experimental data : Internuclear distances, bond angles, vibration frequencies and even chemical reactivity [64,65], or their theoretical counterparts coming from quantum-mechanical calculations in terms of standard s, p, d … atomic contributions. We are not willing to go back to this matter in its entirety, because it is presented in textbooks for most of these properties ; as little less standard topics, we have preferred to turn us to those properties which are known by the generic name of spin coupling constants in magnetic resonance spectroscopy.
The quantum-mechanical picture of hyperfine structures presented by the spin-spin nuclear magnetic resonance (NMR) and electron-spin resonance (ESR) spectra involves a variety of spin Hamiltonian parameters of molecular origin whose magnitude determines that of the coupling constants. In such an analysis, the most characteristic term arises from the Fermi -or contact - operator:
(see e.g. [66]), the importance of which is connected with the values of electron densities (in NMR) or spin densities (in ESR) on the nuclei experimentally tested. The specific form of Fermi operators implies that the magnitude of the contact terms is determined by the s local character of the molecular wave function on some nuclei, i.e., by the relative importance of its s atomic components in these points. Let us add that other spin operators involved in magnetic resonance spectroscopy, such as the electron-spin and spin-spin dipole operators give, for their part, contributions determined by the p character of the wave function. In this matter, hybridization is of paramount importance for the understanding of spin coupling constants in terms of electronic structure.
The 1JXY coupling constants between two chemically bonded nuclei X and Y in NMR give a striking example of the explanatory power of the hybridization concept, especially for the 13C -H and 13C - 13C couplings, where the Fermi contact term is known to be dominant over all the other types of magnetic interactions. Their experimental values can be rationalized by formulas of the form:
k and ℓ being parameters determined from spectroscopic data by least-square fittings in appropriate series of chemical compounds, and s the s-percentage of the atomic hybrids forming the X - Y bond. Actually, this relationship works well for the couplings across the CH and CC bonds of acetylene, ethylene and ethane, where the s fractional character of the sp, sp2 and sp3 standard hybrids varies from 0.50 to 0.33 and 0.25. Of course, explicite hybridization calculations are needed for hydrocarbons having particular structures (for instance three- or four-membered cycles [67)]). The k proportionality factor is much greater than the ℓ constant, so that the latter may be ascribed to small perturbation effects not included in the Fermi contact term.
In ESR spectroscopy, the large quantity of data gathered between 1960 and 1980 has been interpreted in terms of σ and π spin densities, according to the orbital description of the unpaired electron in an independent-particle model. Within this picture, a σ radical has a singly occupied orbital belonging to the same subset as the hybridized bond and lone-pair σ orbitals, while a π radical uses an orbital built from atomic functions perpendicular to the planar or locally planar σ framework [68,69]. So it is easy to understand by symmetry reasons why the electron-spin coupling constants of the σ free radicals are dominated by the direct contribution of the contact term, the magnitude of which depends on the s character of the singly occupied orbital, whereas the couplings of the π radicals have indirect contributions due to σ–π spin-polarisation mechanisms only [70,71].
Still, the concept of hybridization is relevant even for electron-proton couplings aiH because their values are practically determined by the spin density ρi located on the next-neighbour atom i of the proton considered, for instance the unsaturated carbon C to which H is attached. These couplings are governed by relationships of the form
where Q is a proportionality factor possibly depending on other characteristics of carbon Ci. If the spin densities are known, it is possible to derive an empirical value Qexp for this factor using experimental data. The variation of Qexp inside a series of related molecules enables us, by comparison with the bond angles, to see whether the usual hypothesis of atomic hybrids directed to each other has to mitigate in some cases.
To illustrate the last point, we will consider the ESR spectra coming from an unpaired electron in π electron systems of unsaturated monocycles CnHn. Using 1/n as spin densities ρiπ for carbon Ci , the ratio Qexp = aH/ ρiπ computed from the experimental data does not change appreciably, in spite of apex angles CCC deviating very much from the 120° standard value for n = 6. This fact is easily understood in the frame of the hybridization theory by assuming that Qexp is not determined by the values of the cyclic bond angles, but by the less sensitive values predicted by the Del Re hybridization theory [72] ; in other words, the straight bond hypothesis has to be relaxed for strongly strained systems.
Let add that similar considerations cannot be developed without caution in the case of long-range spin parameters of NMR and ESR spectroscopies, because these couplings result from a delicate balance of many effects (see [73]). Their study (including possible vibrational contributions) requires much sophistification in the application of quantum mechanical methods to the evaluation of spin-hamiltonian parameters by perturbation theories at the semiempirical level (see e.g. [74]) as well as at the non-empirical one. A striking example in ESR spectroscopy is the calculation of the electron-proton spin coupling constants for vinyl, the prototype of σ free radicals [75].
3.3 Quantum-chemical flashback
Beside its various applications to structural purposes in Chemistry, the concept of hybridization has a long story in Theoretical Chemistry as an intermediate tool in the construction of realistic molecular wave functions. In the old days of Quantum Mechanics, it was confidently used to determine bond and lone-pair functions for molecular orbital and valence-bond treatments, especially in the frame of the perfect pairing approximation (see e.g. [5]). Later, this approach was revived in order to start with convenient building blocks for writing a total wave function where electronic interaction can be introduced either in a perturbative way, as in the PCILO method [76], or in a variational way passing through quasi-localized orbitals at various approximation levels of the VB and MO theories, (for a recent example see [77]). Presently, SCF-CI treatments using delocalized molecular orbitals seem to be still preferred, but it may be interesting for many reasons including Chemistry to have the possibility of replacing a set of pure s,p,d etc… functions by its equivalent in terms of hybrids adapted to the structure of the molecule in question. This is the aim of hybridization procedures, like the Del Re method. Both points of view have been exploited in the CS-INDO-CI semi-empirical method [78,79], where the basic core parameters responsible for the agreement with experiment as concerns conformations and spectra are determined on a set of hybrid orbitals, but the CI computation of the electronic spectrum itself uses pure atomic orbitals, just for convenience’s sake.
To conclude this review, we should like emphasize the fact that no serious argument can be presented for an exclusive use of pure atomic orbitals in quantum-chemical calculations, except that of the separation of the radial and angular parts of the wave function in the Hartree-Fock picture of the atoms themselves [80]. To the defenders of the traditional s, p, d … orbitals, we wish to reply that there are four coordinate systems for which the Schrödinger equation of the hydrogenic atom can be solved [81], instead of eleven for a wave equation without any potential term (see e.g. [82,83]), and that one of them, the parabolic system, gives rise to sp hybrids directly [84]. Taking this argument stricto sensu , we could be tempted to connect the origin of hybridization to the degeneracy of the s and p levels in hydrogen-like atoms ; and so its importance should gradually decrease from carbon where the 2s and 2p levels have very close orbital energies to nitrogen and oxygen atoms, in conformity with the primitive ideas of Pauling. However, we can also state that the formation of a chemical bound creates an internal electric field suggesting by its analogy to a Stark effect that the polar-coordinates picture is not really adequate for atoms in molecules. More generally, hybridization can be considered as a way to prepare a local set of orbitals belonging to some irreducible representations of a certain point group [19], in other words to break the isotropy of the atom and to raise the organization level of matter in this way[85].