
Solvent-mediated proton transfer reactions in cytosine: an ab initio study
Simone Morpurgo*; Mario Bossa; Giorgio O. Morpurgo Dipartimento di Chimica, Università degli Studi di Roma “La Sapienza”, P.le A. Moro 5, 00185 Roma, Italy
* Corresponding author
Abstract
Solvent-mediated proton transfer reactions of mono- and dihydrated cytosine tautomers were investigated by ab initio quantum chemical calculations. MP2/6-31G** and MP2/D95** energy calculations were performed over geometries obtained at the HF/3-21G level. Corrections for the zero-point vibrational energy were included. It is shown that the coordination of one or two water molecules to cytosine tautomers produces a relative stabilization consistent with that obtained by continuum-based methods (PCM, SCRF). One or two water molecules are however insufficient to completely reproduce the order of stability. Coordination of even a single water molecule strongly activates the proton transfer process with respect to the gas phase. When corrections for ZPE are included in the calculations, water-promoted tautomerization reactions of cytosine have activation energies ranging from 10 to 16 kcal mol− 1 and are therefore expected to occur at room temperature.
1 Introduction
The relative stability of the tautomers of purine and pyrimidine bases is of fundamental importance to the structure and functioning of nucleic acids. The occurrence of rare tautomers was considered a factor responsible for the formation of mismatches leading to spontaneous mutations in the genetic code [1,2]. Cytosine, in particular, has been the subject of several studies, both experimental [3–5] and theoretical [5–15] which have provided a reliable picture of the relative stability of its tautomers, both in the gas phase and in solution. Tautomerization is generally the result of proton transfer (PT) reactions whose activation barriers may exert a kinetic control over the formation of some tautomers. As far as cytosine is concerned, a large majority' of the studies available in the literature focus on the thermodynamic aspects of tautomerization and quite a few [16–19] are devoted to the elucidation of the kinetic aspects. The tautomerization of cytosine in the gas phase, with a special attention to the activation energy of the proton transfer reactions, has been afforded by this group in a previous paper [19]. By comparison with experimental data [4,5] it was shown that kinetic barriers in the gas phase are generally high (30-37 kcal mol− 1) and may affect the tautomers’ population at T ≤ 500 K. On the other hand, it is well known that the distribution of tautomeric forms, especially when it is proton transfer dependent, is modified under the influence of the environment. In this context, two factors are important the effect of the environment (i) on the relative stability of each tautomer and (ii) on the barrier height along the reaction coordinate separating two tautomeric forms.
The effect of the solvent is usually modelled either by the use of the Onsager’s self consistent reaction field (SCRF) [20] or by the polarizable continuum method (PCM) [21]. With regard to the relative stability of cytosine tautomers in aqueous solution, these methods provided results [14,15] which, in spite of some discrepancies, are in reasonable agreement with experimental data [3]. However, continuum-based methods do not explicitly take into consideration the local solvent-solute interaction which is instead important in the description of the proton transfer mechanism in hydrogen-bonded systems. A reasonable approach to the problem was recently proposed [22,23] in which the molecule of interest and few solvent molecules are treated as a supermolecule acting as solute, while the bulk of the solvent is represented as a polarizable dielectric.
In the present paper the thermodynamic and kinetic aspects of the proton transfer reactions among cytosine tautomers assisted by specific solvent molecules was theoretically investigated. For the time being, bulk solvent effects were not considered and attention was only focused on the influence of hydrogen bonding on both (i) tautomers’ relative stability and (ii) the catalytic process occurring between adjacent positions of cytosine. The computational results on point (i) are compared with those of PCM calculations [15]. The results on point (ii) are discussed with reference to the conclusions of other theoretical studies available in the literature [16,17].
2 Theoretical Methods
The Gaussian 94 W [24] program package was employed for the ab initio calculations. The geometries of both minima and transition states were optimized at the HF/3-21G level using the Berny algorithm [25,26]. The transition states were localized by means of the STQN method [26,27]. All structures were vibrationally characterized, checking for the absence of imaginary frequences in the minima and for the presence of only one imaginary frequency in the transition states. It was additionally verified through the graphical interface of HyperChem 4.5 [28] that the imaginary frequency of the transition states corrisponds to the reaction coordinate of the proton transfer reaction. Zero-point vibrational energies (ZPE) were scaled by 0.91, which is a commonly accepted correction factor [5,9]. Single-point energy calculations were performed at the MP2/6-31G** and MP2/D95** level, the latter basis set being employed only for the monohydrated adducts (see next section). The frozen-core (FC) approximation was adopted for all MP2 calculations with only few exceptions, where full MP2 correlation and D95** basis set were additionally used for the sake of comparison with literature data [17].
3 Results and discussion
3.1 Systems investigated
the five most stable tautomers of cytosine are shown in Fig. 1. The tautomeric equilibrium among these species is based on the C1 ![]() C2, C1
C2, C1 ![]() C3 and C4
C3 and C4 ![]() C5 interconversions. They may occur as direct proton transfer reactions in the gas phase, with activation energies ranging from 30 to about 37 kcal mol− 1, as suggested in a previous paper [19].
C5 interconversions. They may occur as direct proton transfer reactions in the gas phase, with activation energies ranging from 30 to about 37 kcal mol− 1, as suggested in a previous paper [19].
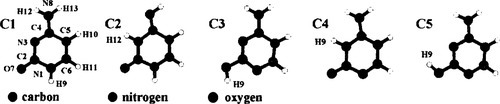
For mono- and dihydrated adducts of cytosine the N1, N3, O7 and N8 positions were considered for coordination, since these sites are the most suitable to hydrogen bind solvent molecules, as already shown both theoretically [16,29] and experimentally on molecules modelling cytosine and isocytosine [30–33]. H10 and H11 were expected to form comparatively weaker hydrogen bonds with solvent molecules and thus coordination to these sites was not considered. The water-assisted reactions investigated in the present study may be ideally divided into three groups (Fig. 2). The first one (reactions a, b, c) encloses the processes catalyzed by one hydrogen-bonded water molecule, which forms a bridge between the two adjacent sites involved in the proton transfer. The second group (d, e, f) deals with the same reactions, catalyzed by two bridging water molecules which give rise to a concerted triple proton transfer mechanism. The mechanisms considered in the third group (g, h, i, l) are the same as those in the first one, with a second water molecule coordinated to some other functional groups of cytosine not directly involved in the proton transfer. The C3 ![]() C5 interconversion, which takes place in the gas phase by rotation of O7-H9 bond around the C2-O7 axis with an activation energy of about 8 kcal mol− 1 [19], in water solution is more likely to occur through a dissociative mechanism and therefore was not included in the present work.
C5 interconversion, which takes place in the gas phase by rotation of O7-H9 bond around the C2-O7 axis with an activation energy of about 8 kcal mol− 1 [19], in water solution is more likely to occur through a dissociative mechanism and therefore was not included in the present work.

The structures reported in Fig. 3 are not directly involved in proton transfer reactions (some of them would eventually give rise to the rather unstable imino-hydroxy tautomeric forms [6,15]) but were included in the calculations in order to investigate the relative stability of mono- and dihydrated cytosine tautomers. The computational results are presented and discussed in three separate sections which are respectively dedicated (3.2) to the effect of hydrogen bonding on the geometrical parameters of the solvated molecules, (3.3) to the influence of hydration on the thermodynamic stability of cytosine tautomers and (3.4) to the kinetics of solvent-assisted proton transfer reactions.

3.2 Geometrical parameters
the main HF/3-21G geometrical parameters of monohydrated cytosine tautomers and transition states are reported in Table 1. For the sake of comparison, the geometries of the C1-C4 tautomers calculated in vacuum at the same computational level are available in Ref. [6]. The structural changes induced in the solute by hydrogen bonding with one solvent molecule are generally consistent with those calculated at higher computational level for some related systems [22,23,34]. As an example, the hydrogen bond at N3 and H12 in the C1-1a adduct induces the following changes with respect to the geometry optimized in vacuum, (i) r(N8-H12) is increased by 0.017 Å, (ii) r(C4-N8) is shortened by 0.013 Å and (iii) r(N3-C4) increases by 0.017 Å. This implies that, as H12 tends to be moved away from N8, the double bond character of C4-N8 increases and, at the same time, the double bond character of N3-C4 decreases. As a further example, in the C1-1b adduct, hydrogen bonding (i) increases both r(N1-H9) and r(C2-O7) by 0.019 Å and (ii) decreases r(N1-C2) by 0.015 Å. The structural changes calculated for the adducts of the other tautomers are quantitatively similar. It should be stressed that geometry optimization at the HF/3-21G level tends to underestimate hydrogen bond distances with respect to the values obtained by using more extended basis sets with inclusion of polarization functions and treatment of electron correlation. However, it will be shown in the 3.3 section that, in the present case, such inaccuracies are not likely to affect appreciably the calculated values of the proton transfer activation energy.
Table 1
Selected geometric parameters (distances in Å, angles in degrees) for monohydrated cytosine tautomers and transition states optimized at the HF/3-21G level.
| C1-1a | TS12-1a | C2-1a | C1-1b | TS13-1a | C3-1a | C4-1a | TS45-1a | C5-1a | |
| r(2-1) | 1.411 | 1.403 | 1.387 | 1.400 | 1.371 | 1.340 | 1.363 | 1.334 | 1.322 |
| r(3-2) | 1.366 | 1.359 | 1.363 | 1.358 | 1.332 | 1.322 | 1.407 | 1.378 | 1.342 |
| r(4-3) | 1.313 | 1.355 | 1.388 | 1.305 | 1.322 | 1.328 | 1.345 | 1.333 | 1.333 |
| r(5-4) | 1.443 | 1.445 | 1.459 | 1.436 | 1.419 | 1.409 | 1.377 | 1.394 | 1.400 |
| r(6-5) | 1.337 | 1.335 | 1.327 | 1.342 | 1.356 | 1.364 | 1.396 | 1.378 | 1.371 |
| r(1-6) | 1.355 | 1.359 | 1.371 | 1.348 | 1.337 | 1.339 | 1.306 | 1.325 | 1.333 |
| r(7-2) | 1.213 | 1.212 | 1.212 | 1.230 | 1.282 | 1.322 | 1.229 | 1.280 | 1.322 |
| r(8-4) | 1.331 | 1.298 | 1.269 | 1.342 | 1.341 | 1.344 | 1.346 | 1.346 | 1.349 |
| r(9-l) | 0.998 | 0.998 | 0.997 | 1.017 | 1.290 | 1.910 | |||
| r(9-3) | 1.020 | 1.280 | 1.942 | ||||||
| r(12-8) | 1.014 | 1.231 | 1.875 | 0.998 | 0.997 | 0.997 | 0.997 | 0.996 | 0.996 |
| r(13-8) | 0.995 | 0.999 | 1.008 | 0.994 | 0.995 | 0.994 | 0.995 | 0.995 | 0.995 |
| r(14-9) | 1.791 | 1.199 | 0.982 | 1.763 | 1.207 | 0.979 | |||
| r(14-12) | 1.817 | 1.258 | 0.984 | ||||||
| r(15-3) | 1.903 | 1.222 | 1.019 | ||||||
| r(15-7) | 1.806 | 1.212 | 0.993 | 1.809 | 1.223 | 0.991 | |||
| r(15-14) | 0.983 | 1.270 | 1.796 | 0.982 | 1.210 | 1.647 | 0.983 | 1.199 | 1.657 |
| r(16-14) | 0.966 | 0.965 | 0.966 | 0.965 | 0.963 | 0.965 | 0.965 | 0.963 | 0.966 |
| a(14-9-1) | 146.75 | 147.79 | 138.66 | ||||||
| a(14-9-3) | 148.84 | 148.78 | 138.95 | ||||||
| a(14-12-8) | 148.88 | 149.97 | 146.30 | ||||||
| a(14-15-3) | 145.48 | 149.81 | 149.42 | ||||||
| a(14-15-7) | 146.57 | 152.69 | 157.25 | 145.53 | 152.43 | 157.54 | |||
| a(15-14-9) | 82.99 | 87.29 | 86.75 | 83.07 | 86.79 | 86.06 | |||
| a(15-14-12) | 82.11 | 85.55 | 82.21 | ||||||
| a(16-14-15) | 108.68 | 117.39 | 115.93 | 109.21 | 118.31 | 118.98 | 109.24 | 117.31 | 116.95 |
| d(16-14-15-9) | 122.94 | 122.47 | 111.49 | -125.04 | -122.43 | -110.78 | |||
| d(16-15-14-12) | -119.91 | -120.13 | -107.68 |

Hydrogen bond interactions are also reflected in the calculated IR spectra of the monohydrated bases. Taking the C1-1a adduct as an example, the following shifts are observed with respect to the frequencies of the isolated C1 tautomer [5,9] (all values are scaled by 0.91): (i) -40 cm− 1 for the -NH2 asymmetric strectching, (ii) -321 cm− 1 for the O14-H15 stretching, (iii) -34 cm− 1 for the O14-H16 stretching and (iv) + 67 cm− 1 for the -NH2 scissoring. Such frequency shift values are in reasonable agreement with those observed in Ar matrices and theoretically calculated for strictly related systems, such as monohydrated 4-aminopyrimidine [31] and 1-methylcytosine [32]. Furthermore, the shift of the C = O stretching mode (-24 cm− 1) calculated for the C1-1b adduct is close to the value experimentally observed for either 1- and 3-methyluracile (ca. -20 cm− 1) [35].
3.3 Relative stability of tautomeric forms
it is widely accepted, on the grounds of both experimental [4,5] and theoretical [8–15] work, that the order of stability of cytosine tautomers in the gas phase is C3 > C5 > C1 > C2 > > C4, the first four tautomers being enclosed in a range of about 3 kcal mol− 1 and C4 being less stable than C3 by more than 7 kcal mol− 1. A rather different ordering exists in polar solvents, where C1 and C4 are the only experimentally observed species [3]. This finding was substantially confirmed by theoretical calculations. It was established, by either the SCRF and the PCM methods, that C1 is the most stable form in aqueous solution and it is preferred by about 5 kcal mol− 1 over the other tautomers, which are enclosed in a range of 1.5 kcal mol− 1 [14,15]. Notably, the C4 tautomer, which is energetically unfavoured in vacuum, becomes the second stable form in aqueous solution, being stabilized by its high dipole moment. Since continuum-based computational methods deal with a single component of the solvation process, namely the dipolar interaction, the present work was also aimed at elucidating how the formation of specific hydrogen bonds with solvent may affect the relative stability of the tautomeric forms of a selected species. Cytosine has several sites which are potentially capable of hydrogen bonding and, therefore, it appears as a particularly interesting case study.
The relative stability of cytosine tautomeric forms was estimated in the following way (i) the energy of all adducts was calculated with inclusion of ZPE, (ii) the most stable adduct with a given number of solvent molecules was identified among all tautomers, (iii) the energy of all other adducts was related to the energy of the most stable one. The results are listed in Table 2. At the MP2(FC)/6-31G**//HF/3-21G level, the most stable monohydrated adducts were found to be C1-1b, C2-1b, C3-1a, C4-1a and C5-1a, and the most stable dihydrated ones C1-2c, C2-2b ~ C2-2c, C3-2b, C4-2a and C5-2a respectively (Figs. 2-3). No appreciable differences were found when the ordering of monohydrated adducts was evaluated at the MP2(FC)/D95**//HF/3-21G level. The trend of the energy difference between the tautomeric forms when an increasing number of water molecules are coordinated to the bare cytosine is shown in Fig. 4. The mean energy of each mono- or dihydrated tautomeric species, ![]() , is calculated according to the Maxwell-Boltzmann distribution law
, is calculated according to the Maxwell-Boltzmann distribution law
![]()
Table 2
Computational results obtained for the adducts of Figures 2-3. Energy (a.u), dipole moment (Debye), zero-point vibrational energy (ZPE/kcal mol− 1), ZPE-corrected relative energy of the adducts (ΔErel/kcal mol− 1) with stability order in brackets (a), activation energy uncorrected (ΔE‡/kcal mol− 1) and corrected (ΔE‡corr/kcal mol− 1) forZPE.
| MP2(FC)/6-3 lG**//HF/3-21G results | ||||||
| monohydrated adducts | ||||||
| adduct | energy | dipole | ZPE(b) | ΔErel | ΔE‡ | ΔE‡Corr |
| formamide | -245.67403 | 3.32 | 44.07 | 0.00 | 23.65 | 20.33 |
| TS | -245.63634 | 2.72 | 40.75 | |||
| formamidic ac. | -245.65512 | 2.30 | 43.89 | 11.69 | 11.78 | 8.64 |
| formamidinc | -225.81646 | 3.21 | 51.43 | 18.93 | 15.84 | |
| TS | -225.78629 | 4.19 | 48.34 | |||
| C1-1a | -470.04835 | 6.74 | 77.02 | 0.89 | 18.51 | 15.04 |
| TS12-1a | -470.01886 | 6.89 | 73.55 | |||
| C2-1a | -470.04476 | 6.13 | 77.25 | 3.37 | 16.25 | 12.55 |
| C1-1b | -470.04958 | 6.14 | 76.90 | 0.00 (1) | 15.24 | 11.43 |
| TS13-1a | -470.02530 | 5.46 | 73.09 | |||
| C3-1a | -470.04844 | 4.58 | 76.37 | 0.19 (2) | 14.52 | 11.24 |
| C4-1a | -470.03968 | 7.38 | 76.64 | 5.95 (5) | 13.13 | 9.39 |
| TS45-1a | -470.01875 | 7.06 | 72.90 | |||
| C5-1a | -470.04524 | 6.22 | 76.22 | 2.04 (3) | 16.62 | 13.30 |
| C2-1b | -470.04534 | 4.01 | 77.17 | 2.93 (4) | ||
| C2-1c | -470.04163 | 5.41 | 76.98 | 5.07 | ||
| C3-1b | -470.04785 | 2.74 | 76.40 | 0.59 | ||
| C5-1b | -470.04374 | 3.76 | 76.29 | 3.05 | ||
| C1-1a (c) | -470.04631 | 5.81 | 76.51 | 20.90 | 17.29 | |
| TS12-1a (c) | -470.01300 | 6.15 | 72.90 | |||
| C2-1a (C) | -470.04198 | 5.20 | 76.65 | 18.19 | 14.44 | |
| dihydrated adducts | ||||||
| C1-2a | -546.28996 | 6.16 | 92.30 | 0.82 | 18.83 | 14.06 |
| TS12-2a | -546.25995 | 6.34 | 87.53 | |||
| C2-2a | -546.28449 | 5.59 | 92.48 | 4.44 | 15.40 | 10.45 |
| C1-2b | -546.29073 | 5.26 | 92.23 | 0.27 | 1638 | 11.38 |
| TS13-2a | -546.26463 | 3.88 | 87.23 | |||
| C3-2a | -546.28654 | 3.90 | 91.44 | 2.11 | 13.75 | 9.54 |
| C4-2a | -546.28574 | 6.14 | 92.48 | 3.65 (5) | 15.11 | 10.22 |
| TS45-2a | -546.26166 | 5.30 | 87.59 | |||
| C5-2a | -546.28667 | 5.19 | 91.82 | 2.41 (3) | 15.69 | 11.46 |
| C1-2c | -546.29156 | 5.26 | 92.48 | 0.00 (1) | 18.99 | 15.43 |
| TS12-2b | -546.26129 | 5.25 | 88.92 | |||
| C2-2b | -546.28630 | 4.44 | 92.61 | 3.43 | 15.69 | 12.00 |
| C1-2c | -546.29156 | 5.26 | 92.48 | 0.00 (1) | 15.66 | 11.81 |
| TS13-2b | -546.26660 | 4.46 | 88.63 | |||
| C3-2b | -546.28888 | 3.60 | 91.87 | 1.07 (2) | 13.98 | 10.74 |
| C1-2d | -546.28468 | 8.66 | 91.50 | 3.34 | 15.84 | 12.04 |
| TS13-2c | -546.25943 | 7.80 | 87.70 | |||
| C3-2c | -546.28134 | 6.65 | 90.86 | 4.79 | 13.75 | 10.59 |
| C4-2b | -546.27484 | 10.84 | 91.26 | 9.27 | 13.64 | 9.93 |
| TS45-2b | -546.25311 | 10.48 | 87.55 | |||
| C5-2b | -546.27817 | 9.51 | 90.70 | 6.62 | 15.73 | 12.58 |
| C2-2c | -546.28628 | 3.20 | 92.57 | 3.40 (4) | ||
| C2-2d | -546.28070 | 4.78 | 92.33 | 6.66 | ||
| C3-2d | -546.28790 | 1.86 | 91.67 | 1.49 | ||
| C5-2c | -546.28649 | 4.26 | 92.04 | 2.74 | ||
| MP2(FC)/D95**//HF/3-2IG results | ||||||
| monohydrated adducts | ||||||
| formamide | -245.76834 | 3.46 | 44.07 | 0.00 | 22.48 | 19.16 |
| TS | -245.73251 | 2.80 | 40.75 | |||
| Fonnamidic ac. | -245.75031 | 2.33 | 43.89 | 11.13 | 1117 | 8.03 |
| formamidinc | -225.90205 | 3.32 | 51.43 | 17.70 | 14.61 | |
| TS | -225.87384 | 4.28 | 48.34 | |||
| C1-1a | -470.12338 | 6.87 | 77.02 | 0.45 | 17.72 | 14.25 |
| TS12-1a | -470.09514 | 6.98 | 73.55 | |||
| C2-1a | -470.11910 | 6.21 | 77.25 | 3.37 | 15.04 | 11.34 |
| C1-1b | -470.12391 | 6.25 | 76.90 | 0.00 (1) | 14.43 | 10.62 |
| TS13-1a | -470.10092 | 5.56 | 73.09 | |||
| C3-1a | -470.12263 | 4.66 | 76.37 | 0.27 (2) | 13.62 | 10.34 |
| C4-1a | -470.11428 | 7.53 | 76.64 | 5.78 (5) | 12.35 | 8.61 |
| TS45-1a | -470.09460 | 7.19 | 72.90 | |||
| C5-1a | -470.11944 | 6.32 | 76.22 | 2.12 (3) | 15.59 | 12.27 |
| C2-1b | -470.11947 | 4.09 | 77.17 | 3.06 (4) | ||
| C2-1c | -470.11554 | 5.50 | 76.98 | 5.33 | ||
| C3-1b | -470.12238 | 2.82 | 76.40 | 0.46 | ||
| C5-1b | -470.11797 | 3.87 | 76.29 | 3 12 | ||
| C1-1a (d) | -470.25632 | 6 87 | 77.02 | 17.81 | 14.34 | |
| TS12-1a (d) | -470.22793 | 6.98 | 73.55 | |||
| C2-1a (d) | -470.25204 | 6 21 | 77.25 | 15.13 | 11.43 | |
| C1-1a (e) | -470.05474 | 6.89 | 77.02 | 19.77 | 16.30 | |
| TS12-1a (e) | -470.02323 | 7.07 | 73.55 | |||
| C2-1a (e) | -470.05002 | 6.27 | 77.25 | 16.81 | 13.11 | |
| C1-1a (f) | -470.12257 | 5.88 | 76.51 | 19.99 | 16.38 | |
| TS12-1a (f) | -470.09072 | 6.22 | 72.90 | |||
| C2-1a (f) | -470.11761 | 5.24 | 76.65 | 16.87 | 13.12 | |
| C1-1a (g) | -470.25553 | 5.86 | 76.51 | 20.07 | 16.46 | |
| TS12-1a (g) | -470.22355 | 6.21 | 72.90 | |||
| C2-1a (g) | -470.25056 | 5.24 | 76.65 | 16.95 | 13.20 | |
| C1-1a (h) | -470.05400 | 5.85 | 76.51 | 22.38 | 18.77 | |
| TS12-1a (h) | -470.01834 | 6.25 | 72.90 | |||
| C2-1a (h) | -470.04858 | 5.26 | 76.65 | 18.98 | 15.23 | |


(a) the most stable adduct is considered for each tautomeric form;
(b) calculated at the HF/3-21G level and scaled by 0.91 [5,9];
(c) MP2(FC)/6-31 G**//HF/3-21G, Cs constraint;
(d) MP2(full)/D95**//HF/3-21G;
(e) MP2(FC)/D95*//HF/3-21G;
(f) MP2(FC)/D95**//HF/3-21G, Cs constraint;
(g) MP2(full)/D95**//HF/3-21G, Cs constraint;
(h) MP2(FC)/D95*//HF/3-21G, Cs constraint.

where the summation is over the adducts of a given tautomer and Ei is the relative energy with respect to the most stable adduct of all tautomers considered [point (iii) above]. The values at the bottom of the figure are those calculated for unsolvated cytosine monomers [19]. The values at the top are those calculated by Colominas et al. [15] by means of the PCM method. It is apparent that two water molecules are insufficient to account for the final order of stability reported in [15], though some partial results are achieved: (i) C1, which is the third most stable form in vacuum (+ 1.61 kcal mol− 1 with respect to C3) becomes the most stable form upon addition of one water molecule (-0.16 kcal mol− 1 with respect to C3) and the stabilization is enhanced (-1.09 kcal mol− 1) as a second water molecule is coordinated, (ii) the stabilization of C4 which, being less stable than C3 by about 8.5 kcal mol− 1 in vacuum, becomes less stable than C1 by about 3.3 kcal mol− 1 when the dihydrated adducts are considered.
3.4 Kinetics of water-assisted proton transfer
as anticipated in Section 3.2, HF/3-21G geometry optimizations tend to underestimate hydrogen bond distances with respect to the values obtained at higher computational level. As a consequence, MP2/6-31G** single-point calculations based on HF/3-21G geometries may yeld, in principle, inaccurate values for the PT activation energies. For this reason, we have performed benchmark calculations on the water-assisted tautomerization equilibria of formamidine and formamide ![]() formamidic acid. Such systems were chosen as a reference because the proton transfer occurs between the same functional groups as those involved in cytosine. The results of the computations at the MP2(FC)/6-31 G**//HF/3-21G and MP2(FC)/D95**//HF/3-21G level are reported in Table 2. Comparison with MP2/6-31G**//MP2/6-31G** literature data [22,34] shows that the activation energies are computed with an error of about ± 1.5 kcal mol− 1. On the assumption that this approximation can be extended to proton transfer reactions of cytosine, we are led to conclude that HF/3-21G geometiy optimizations are adequate for the scope of the present work.
formamidic acid. Such systems were chosen as a reference because the proton transfer occurs between the same functional groups as those involved in cytosine. The results of the computations at the MP2(FC)/6-31 G**//HF/3-21G and MP2(FC)/D95**//HF/3-21G level are reported in Table 2. Comparison with MP2/6-31G**//MP2/6-31G** literature data [22,34] shows that the activation energies are computed with an error of about ± 1.5 kcal mol− 1. On the assumption that this approximation can be extended to proton transfer reactions of cytosine, we are led to conclude that HF/3-21G geometiy optimizations are adequate for the scope of the present work.
The results obtained for the proton transfer reactions of cytosine illustrated in Fig. 2, are listed in Table 2. The activation energies are reported as a simple difference in electronic energy and with further correction for ZPE. As it is generally observed for PT reactions [22,34,36], the inclusion of ZPE lowers the activation barriers by 2-4 kcal mol− 1 with respect to the values calculated on the basis of pure electronic energy changes. As ZPE was considered, it was found that the activation energies of C1 ![]() C3 and C4
C3 and C4 ![]() C5 direct and reverse processes range from 10 to 12 kcal mol− 1, with small differences due to the relative energy of the minima. The C1 → C2 tautomerization has an activation energy of about 14-15 kcal mol− 1, while the reverse reaction, C2 → C1, has an activation energy of ca. 12 kcal mol− 1. For the processes labelled as a, b and c MP2(FC)/D95** single point energy calculations produced activation energies which are, on the average, 0.92 kcal mol− 1 lower than those calculated at MP2(FC)/6-31G** level.
C5 direct and reverse processes range from 10 to 12 kcal mol− 1, with small differences due to the relative energy of the minima. The C1 → C2 tautomerization has an activation energy of about 14-15 kcal mol− 1, while the reverse reaction, C2 → C1, has an activation energy of ca. 12 kcal mol− 1. For the processes labelled as a, b and c MP2(FC)/D95** single point energy calculations produced activation energies which are, on the average, 0.92 kcal mol− 1 lower than those calculated at MP2(FC)/6-31G** level.
Remarkably, the inclusion of even a single bridging water molecule in the mechanism reduced the activation energy by 15-20 kcal mol− 1 in comparison to the direct tautomerization in the gas phase [19]. This result agrees with those of recent investigations on some related systems such as the tautomeric couples formamide/formamidic acid [22], 2-hydroxypyridine/2-pyridone [23], formamidine [34] and others [37,38], where a comparable lowering of the activation energy was found for the water-assisted reaction with respect to the gas-phase process. Notably, the major kinetic effect is produced by the first bridged solvent molecule (reactions a, b, c). Neither the inclusion of a second bridged water molecule (cases d, e, f) nor the coordination of a second water molecule to a site not involved in the proton transfer (cases g, h, i, l) did result in a further kinetic promotion.
The kinetics of the Cl ![]() C2 interconversion mediated by a single water molecule was recently investigated in the ground state as well as in the lowest excited ππ* and nπ* singlet states [17]. In this work, as far as the ground state reaction is concerned, the 3-21G basis set was employed for geometry optimization at the HF level. The minima and the transition states of the cytosine-H2O adduct were optimized (i) without geometrical constraints and (ii) imposing Cs symmetry. In each case, MP2(full)/D95** single point energy calculations were performed along the reaction coordinate. For the C1 → C2 reaction, the authors reported an activation energy of 1.0 eV under the Cs constraint which decreased by about 10% when geometry relaxation was allowed. On this basis, it was suggested that the C1 → C2 proton transfer is extremely unlikely in the ground state, due to its high activation energy. Since our findings disagreed with this conclusion, we were prompted to analyze in deeper detail our computational results. Surprisingly, we were not able to reproduce the results of Ref. [17]. For C1 → C2 we calculated, under the Cs constraint, an activation energy of 20.07 kcal mol− 1 [MP2(full)/D95**] and 19.99 kcal mol1 [MP2(FC)/D95**]. Upon geometry relaxation, these values decreased to 17.81 and 17.72 kcal mol− 1, respectively. We found a relative agreement only by MP2(FC)/D95* single-point calculations, which gave activation energies of 22.38 (Cs) and 19.77 (unconstrained optimization) kcal mol− 1. This result, however, is to be considered physically less reliable, due to the lack of p-type polarization functions on hydrogen atoms in the D95* basis set. Finally, HF/3-21G geometries and energies (-466.03406 a.u. for C1 · H2O and -466.03116 a.u. for C2 · H2O) of the unconstrained minima were found in agreement with those reported in Ref. [17]. A further agreement was found for the energy difference between unconstrained and Cs minima (350 and 480 cm− 1 for C1 · H2O and C2 · H2O, respectively). Unfortunately, the geometry of the transition states was not reported in Ref. [17]. The inclusion of the ZPE, not previously considered, further reduced the activation energy of the C1 → C2 process by about 3.5 kcal mol− 1, to the final value of 14-15 kcal mol− 1. Our results are closer to those of Stepanian et al. [16], although the latter work was performed by the semiempirical MNDO/M [39,40] method and activation energies as low as 12 kcal mol− 1 could only be obtained for the concerted three-proton transfer mechanism.
C2 interconversion mediated by a single water molecule was recently investigated in the ground state as well as in the lowest excited ππ* and nπ* singlet states [17]. In this work, as far as the ground state reaction is concerned, the 3-21G basis set was employed for geometry optimization at the HF level. The minima and the transition states of the cytosine-H2O adduct were optimized (i) without geometrical constraints and (ii) imposing Cs symmetry. In each case, MP2(full)/D95** single point energy calculations were performed along the reaction coordinate. For the C1 → C2 reaction, the authors reported an activation energy of 1.0 eV under the Cs constraint which decreased by about 10% when geometry relaxation was allowed. On this basis, it was suggested that the C1 → C2 proton transfer is extremely unlikely in the ground state, due to its high activation energy. Since our findings disagreed with this conclusion, we were prompted to analyze in deeper detail our computational results. Surprisingly, we were not able to reproduce the results of Ref. [17]. For C1 → C2 we calculated, under the Cs constraint, an activation energy of 20.07 kcal mol− 1 [MP2(full)/D95**] and 19.99 kcal mol1 [MP2(FC)/D95**]. Upon geometry relaxation, these values decreased to 17.81 and 17.72 kcal mol− 1, respectively. We found a relative agreement only by MP2(FC)/D95* single-point calculations, which gave activation energies of 22.38 (Cs) and 19.77 (unconstrained optimization) kcal mol− 1. This result, however, is to be considered physically less reliable, due to the lack of p-type polarization functions on hydrogen atoms in the D95* basis set. Finally, HF/3-21G geometries and energies (-466.03406 a.u. for C1 · H2O and -466.03116 a.u. for C2 · H2O) of the unconstrained minima were found in agreement with those reported in Ref. [17]. A further agreement was found for the energy difference between unconstrained and Cs minima (350 and 480 cm− 1 for C1 · H2O and C2 · H2O, respectively). Unfortunately, the geometry of the transition states was not reported in Ref. [17]. The inclusion of the ZPE, not previously considered, further reduced the activation energy of the C1 → C2 process by about 3.5 kcal mol− 1, to the final value of 14-15 kcal mol− 1. Our results are closer to those of Stepanian et al. [16], although the latter work was performed by the semiempirical MNDO/M [39,40] method and activation energies as low as 12 kcal mol− 1 could only be obtained for the concerted three-proton transfer mechanism.
On the basis of the present calculations on simplified hydrated models we can safely suggest that cytosine tautomerization reactions should take place at room temperature, and that the C1 → C2 conversion is kinetically unfavoured by 2-4 kcal mol− 1 with respect to the C1 ![]() C3 and C4
C3 and C4 ![]() C5 processes. Bulk solvent effects, which are not included in the present study, are expected to provide only a fine tuning of the activation energies, as variations of the dipole moment from minima to transition states are generally less than 1 Debye (Table 2).
C5 processes. Bulk solvent effects, which are not included in the present study, are expected to provide only a fine tuning of the activation energies, as variations of the dipole moment from minima to transition states are generally less than 1 Debye (Table 2).
4 Conclusions
The main computational results of the present work can be summarized as follows: (i) as far as HF/3-21G geometry optimizations are concerned, the structural changes induced in the solute by hydrogen bonding with one solvent molecule are consistent with those calculated at higher computational level for related systems such as monohydrated formamide, formamidic acid and formamidine, but hydrogen bond distances are generally underestimated; (ii) proton transfer activation energies calculated from these geometries through MP2/6-31G** and MP2/D95** single points differ by ca. ± 1.5 kcal mol− 1 from the values based on full MP2/6-31G** optimizations; (iii) coordination of a discrete number of solvent molecules produces a relative stabilization of cytosine tautomers which is close to that obtained by continuum-based methods (PCM, SCRF); (iv) one or two water molecules are however unsufficient to reproduce the final order of stability given in [15]; (v) coordination of even a single water molecule strongly activates the proton transfer process with respect to the gas phase; (vi) coordination of more than one water molecule does not result in a further promotion of the proton transfer reactions; (vii) as ZPE correction is included in the calculations, water-promoted tautomerization reactions of cytosine have activation energies ranging from 10 to 15 kcal mol− 1; (viii) the C1 → C2 conversion (ΔE‡ = 14-15 kcal mol− 1), though kinetically unfavoured with respect to the C1 ![]() C3 and C4
C3 and C4 ![]() C5 processes, can take place at room temperature; (ix) as a general conclusion, kinetic factors do not seem to be important for the distribution of cytosine tautomers in water solution at room temperature. The most stable of the tautomers (C1) is expected to be much less kinetically protected by the solvent than it was suggested in ref. [17].
C5 processes, can take place at room temperature; (ix) as a general conclusion, kinetic factors do not seem to be important for the distribution of cytosine tautomers in water solution at room temperature. The most stable of the tautomers (C1) is expected to be much less kinetically protected by the solvent than it was suggested in ref. [17].