
Validation and regulatory compliance of free/open source software
Abstract:
Open source systems offer a number of advantages, but the need to formally validate some open source applications can be a challenge where there is no clearly defined ‘software vendor’. In these cases the regulated company must assume responsibility for controlling a validated open source application that is subject to ongoing change in the wider software development community. Key to this is knowing which open source applications require validation, identifying the additional risks posed by the use of open source software and understanding how standard risk-based validation models need to be adapted for use with software that is subject to ongoing refinement.
21.1 Introduction
There is no doubt that the use of free/libre open source software (FLOSS) can offer some significant advantages. Within the life sciences industry these include all of the advantages available to other industries such as the ability to use low or no cost software, but the use of open source software can also provide:
![]() the ability to access and use new and innovative software applications in timescales that can be significantly advanced when compared to software developed by commercial vendors;
the ability to access and use new and innovative software applications in timescales that can be significantly advanced when compared to software developed by commercial vendors;
![]() the ability to collaborate with other industry specialists on the development of software to address the general non-competitive needs of interest to the industry.
the ability to collaborate with other industry specialists on the development of software to address the general non-competitive needs of interest to the industry.
As significant changes are taking place within the life sciences industry (such as the move towards greater outsourcing and collaboration, the greater need to innovate and reduce the time to bring products to market), these advantages can be particularly beneficial in areas such as:
![]() the collaborative development of drug candidates by ‘Big Pharma’ working with smaller biotechnology start-ups and or academia (using open source content management and collaborative working) solutions;
the collaborative development of drug candidates by ‘Big Pharma’ working with smaller biotechnology start-ups and or academia (using open source content management and collaborative working) solutions;
![]() the development of innovative medical devices such as pharmacotherapeutic devices or devices using multiple technologies using open source design, modelling and simulation tools (e.g. Simulations Plus [1]);
the development of innovative medical devices such as pharmacotherapeutic devices or devices using multiple technologies using open source design, modelling and simulation tools (e.g. Simulations Plus [1]);
![]() the collaborative conduct of clinical trials including the recruitment of subjects using open source relationship management software (e.g. OpenCRX [2]) or cloud-based social networking tools such as LinkedIn [3];
the collaborative conduct of clinical trials including the recruitment of subjects using open source relationship management software (e.g. OpenCRX [2]) or cloud-based social networking tools such as LinkedIn [3];
![]() the analysis of clinical trial data, the detection of safety signals from adverse events data and the monitoring of aggregated physician (marketing) spend data using open source statistical programming and analysis tools;
the analysis of clinical trial data, the detection of safety signals from adverse events data and the monitoring of aggregated physician (marketing) spend data using open source statistical programming and analysis tools;
![]() the development of process analytical technology (PAT) control schemes, facilitated by open source data analysis and graphical display tools;
the development of process analytical technology (PAT) control schemes, facilitated by open source data analysis and graphical display tools;
![]() the analysis of drug discovery candidate data or manufacturing product quality data, using open source data analysis tools (e.g. for mass spectrometry, see Chapter 4).
the analysis of drug discovery candidate data or manufacturing product quality data, using open source data analysis tools (e.g. for mass spectrometry, see Chapter 4).
21.2 The need to validate open source applications
When such applications have a potential impact on product quality, patient safety or the integrity of regulatory critical data, some of these advantages can be offset by the need to appropriately validate the software.
There is a clear regulatory expectation that such applications are validated – stated in national regulations (e.g. US 21CFR Part 211 [4] and Part 820 [5]), regulatory guidance (e.g. EU Eudralex Volume 4 Annex 11 [6], PIC/S PI-011 [7]) and applicable industry standards (e.g. ISO 13485 [8]). These documents require that such systems are validated, but do not provide detailed requirements on how this is achieved.
Computer system (or software) validation is a process whereby clear and unambiguous user and functional requirements are defined and are formally verified using techniques such as design review, code review and software testing. This should all be governed as part of a controlled process (software development life cycle) defined in a validation plan, supported by appropriate policies and procedures and summarised in a validation report.
Although some regulatory guidance documents (i.e. FDA General Principles of Software Validation [9], and PIC/S PI-011) do provide a high-level overview of the process of computer system validation, industry has generally provided detailed guidance and good practices through bodies such as the International Society for Pharmaceutical Engineering (ISPE) and the GAMP® Community of Practice.
The need to validate such software or applications is not dependent on the nature of the software or how it is sourced – it is purely based on what the software or application does. If an application supports or controls a process or manipulates regulatory significant data that is within the scope of regulations (e.g. clinical trial data, product specifications, manufacturing and quality records, adverse events reporting data) or provides functionality with the potential to impact patient safety or product quality, there is usually a requirement to validate the software.
This also includes an expectation to ensure that the application or software fulfils the users stated requirements, which should, of course, include requirements to ensure compliance with regulations. Validating the software or application therefore provides a reasonably high degree of assurance that:
![]() the software will operate in accordance with regulatory requirements;
the software will operate in accordance with regulatory requirements;
![]() the software fulfils such requirements in a reliable, robust and repeatable manner.
the software fulfils such requirements in a reliable, robust and repeatable manner.
Not all open source software has regulatory significance, but where this is the case failure to validate such software can have serious consequences, which includes regulatory enforcement action. There have been cases where a serious failure to appropriately validate a computerised system has directly led to or has partly contributed to enforcement actions including failure to issue product licences, forced recall of products, import restrictions being imposed or, in the most serious cases, US FDA Consent Decrees [10], which have required life sciences companies to implement expensive corrective and preventative actions and pay penalties in the range of hundreds of millions of dollars for non-compliance.
More importantly, the failure to appropriately validate a computerised system may place product quality and patient safety at risk and there are several known instances where faulty software has led to indirect or direct harm to patients.
21.3 Who should validate open source software?
Although the regulations do not provide details on how software should be validated, those same regulatory expectations (and numerous enforcement actions by various regulatory agencies) make it clear that regulated companies (i.e. pharmaceutical, medical device, biotechnology, biological and biomedical manufacturers, CROs, etc.) are accountable for the appropriate validation of such software applications.
Industry guidance such as the widely referenced GAMP® Guide [11] is increasingly looking for ways in which regulated companies can exercise such accountabilities while at the same time leveraging the activities and documentation of their software vendors or professional services providers (systems integrators, engineering companies, etc.). The GAMP® Guide provides pragmatic good practice for the risk-based validation of computerised systems and is accompanied by a number of companion Good Practice Guides. Although all of these recommended approaches can be applied to the validation of open source software, there is very little specific guidance on the specific validation of such software.
Following one of the key concepts of the GAMP® Guide – leveraging supplier involvement, activities and documentation – presents a significant challenge for the validation of open source applications where there is often no software vendor with which to establish a contractual relationship and often no professional services provider who will assume responsibility for implementing and supporting open source software.
There are examples of third-party organisations providing implementation and support services for open source infrastructure (e.g. Linux Red Hat) or applications. This may not, however, be the case for many new or innovative applications which are at the leading edge of functionality and which may offer significant advantages to a life sciences organisation.
In most cases it is the regulated company which is not only accountable for the validation of the open source application but must also assume day-to-day responsibility for ensuring that the application is regulatory-compliant, for validating the application and for maintaining the application in a validated state with respect to their own operational use of the software. In the case of the same instance of open source software used by multiple regulated companies (e.g. Software as a Service running in a community cloud), it is theoretically possible for a number of regulated companies to form their own open source community and share responsibility for the validated state of the shared software, but this is a relatively new and untested model.
21.4 Validation planning
As with any software or application, the validation process starts with validation planning. Many regulated companies have resources to address this in-house, whereas small to medium organisations may prefer to take the acceptable step of using qualified third parties such as consultants.
Thorough and thoughtful validation planning is the key to successfully validating open source software. The use of open source software is different from the validation of commercially developed and supported applications, and the validation planning should be undertaken by experienced resources that understand and appreciate the differences and can plan the validation accordingly.
21.4.1 Package assessment
Validation planning should start with an assessment of the open source software package (Figure 21.1) and will ask two important questions.
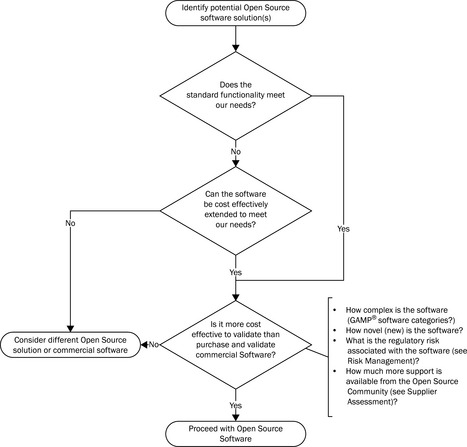
![]() Does (or can) the application deliver functionality that complies with our regulatory requirements?
Does (or can) the application deliver functionality that complies with our regulatory requirements?
![]() Is it possible to cost-effectively validate the open source software in our organisation?
Is it possible to cost-effectively validate the open source software in our organisation?
Like commercial software, open source software can be categorised as GAMP® software category 1, 3, 4 or 5 or a combination thereof (see Table 21.1).
Table 21.1
| GAMP® 5 software category | Description |
| 1 | Infrastructure software (operating system, middleware, database managers, development tools, utilities, etc.) |
| 2 | No longer used in GAMP® 5 |
| 3 | Non-configured software (little/no configurability) |
| 4 | Configured software (configured by the parameterisation of the application for use by the regulated company) |
| 5 | Custom software (custom developed for the regulated company) |
Open source software may come in many forms. At the simplest it may be GAMP® software category 3, with fixed functionality with the possibility to enter only simple parameters. It may be GAMP® software category 4, meaning that it is a configurable off-the-shelf (COTS) application or, because of the nature of open source software it may be customisable – GAMP® software category 5. It may, of course, also combine elements of GAMP® software categories 4 and 5, meaning that a standard configurable application has been functionally extended for specific use by the regulated company or possibly by the wider open source community – the ease with which functionality can be extended is, of course, one of the benefits of open source software.
From a validation perspective, such project-specific development should be considered as customisation (GAMP® software category 5) or at least as novel configurable software (GAMP® software category 4). Categorisation of open source software as GAMP® category 4 or 5 will significantly extend the rigour of the required validation process, which may in turn further erode the cost/benefit argument for using open source software. A question well worth asking is at what point does the further development and validation of an open source software cost more in terms of internal resource time and effort than acquiring an equivalent commercial product?
This is specifically a problem with generic open source software which is not specifically aimed for use in the life sciences industry. An example might be an open source contact management application that is extended to allow sales representatives to track samples of pharmaceutical product left with physicians. In this case the extension to the open source software to provide this functionality may require a significant validation effort, which could cost more than purchasing a more easily validated commercial CRM system designed for the pharmaceutical industry and provisioned as Software as a Service. For software that is specific to the life sciences industry (e.g. an open source clinical trials data entry package), there is always the possibility of further development being undertaken by the wider open source community, which can be a more cost-effective option.
There is then the question of whether the open source software can successfully be validated. Although this usually means ‘Can the software be validated cost-effectively?’, there are examples of open source software that cannot be placed under effective control in the operational environment (see below). This should be assessed early in the validation planning process, so that time and money is not wasted trying to validate an open source application that cannot be maintained in a validated state. This step should also include an initial assessment of the software risk severity (see below) to determine what will be the appropriate scope and rigour of the risk-based validation.
The answer to these questions will determine whether validation is possible and whether it is still cost-effective to leverage an open source solution. Assuming that this is the case, it is then necessary to determine whether it is possible to leverage any validation support from the software ‘supplier’.
21.4.2 Supplier assessment
There is a clear regulatory expectation to assess suppliers. Such assessments (including audits) are potentially subject to regulatory inspection (Eudralex Volume 4, Annex 11). Given the additional risks associated with open source software, it is essential that such an assessment is carefully planned, executed and documented. An overview of the approach is presented in Figure 21.2.
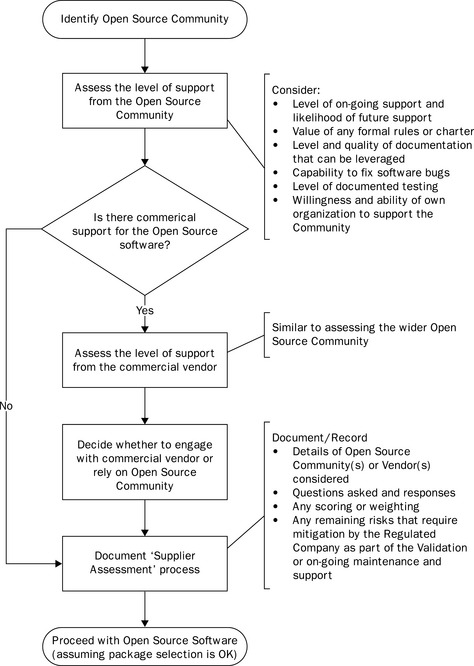
In most cases with open source software there is no supplier to assess, so a traditional supplier audit of a commercial supplier is out of the question. It is, however, possible to assess the support available from the open source community, even if this is not available on a contractual basis.
Key questions to consider and document the answers to include:
![]() Is there a formal community supporting the software or is it just a loose collection of individuals?
Is there a formal community supporting the software or is it just a loose collection of individuals?
![]() Does the community have any formal rules or charter that provide a degree of assurance with respect to support for the software?
Does the community have any formal rules or charter that provide a degree of assurance with respect to support for the software?
![]() How mature is the software? How likely is it that the open source community will remain interested in the development of the software once the immediate development activities are complete?
How mature is the software? How likely is it that the open source community will remain interested in the development of the software once the immediate development activities are complete?
![]() What level of documentation is available within the community? How up-to-date is the documentation compared to the software?
What level of documentation is available within the community? How up-to-date is the documentation compared to the software?
![]() How does the community respond to identified software bugs? Are these fixed in a timely manner and are the fixes reliable?
How does the community respond to identified software bugs? Are these fixed in a timely manner and are the fixes reliable?
![]() What level of testing is undertaken by the community? Is this documented and can it be relied upon in lieu of testing by the regulated company?
What level of testing is undertaken by the community? Is this documented and can it be relied upon in lieu of testing by the regulated company?
![]() What level of involvement are we willing to play in the community? Will we only leverage the software outputs, or actively support the development?
What level of involvement are we willing to play in the community? Will we only leverage the software outputs, or actively support the development?
Although it is not possible to formally audit any supplier, it is possible to consider these questions and form a reasonably accurate set of answers from talking to other regulated companies who are already using the software and by visiting online forums.
In many cases, open source software is used for drug discovery and product development. Examples of such software include general data analytic tools as well as industry-specific bioinformatic and cheminformatic and pharmacogenomic tools (examples include KNIME [12], see Chapter 6, JAS3 [13] and RDKit [14], Bioconductor [15], BioPerl [16], BioJava [17], Bioclipse [18], EMBOSS [19], Taverna workbench [20], and UGENE [21], to name a few among many). Software applications used in drug discovery require no such formal validation, which is just as well as the support from different development communities can vary significantly.
However, in some other cases the level of support is very good and allows such open source software to be used for applications where validation is required. In one example of good practice, the R Foundation [22] not only provides good documentation for their open source statistical programming application, but has also worked in conjunction with compliance and validation experts and the US FDA to:
![]() produce white papers providing guidance on how to leverage community documentation and software tools to help validate their software;
produce white papers providing guidance on how to leverage community documentation and software tools to help validate their software;
![]() provide guidance on how to control their software in an operational environment and maintain the validated state;
provide guidance on how to control their software in an operational environment and maintain the validated state;
![]() address questions on the applicable scope of Electronic Records and Signatures (US 21CFR Part 11 [23]).
address questions on the applicable scope of Electronic Records and Signatures (US 21CFR Part 11 [23]).
Although not solely intended for use in the life sciences industry, these issues were of common concern to a significant number of users within the open source community, allowing people to work together to provide the necessary processes and guidance to support the validation of the software. This level of support has allowed the use of this open source software to move from non-validated use in drug discovery, into validated areas such as clinical trials and manufacturing. In other cases the open source community may only be loosely organised and may have no significant interest in supporting users in the life sciences industry. Although this does not rule out the validation and use of such software, regulated companies should realise that they will incur a significantly higher cost of validation when compared to more organised and supportive communities.
The cost of such validation support needs to be evaluated and this is best achieved by identifying the ‘gaps’ left by the community and estimating the activities, documentation and costs required to initially validate the software and to maintain the validated state. Such gaps should be measured against the requirements identified below.
Following the package assessment, initial risk assessment, supplier assessment and gap analysis, it will be possible to develop a suitable validation plan. This will often be quite unlike the validation plan prepared for a traditional commercial software package and regulated companies need to be wary of trying to use a traditional validation plan template, which may be unsuited to the purpose. It is important that the validation plan reflects the specific nature of and risks associated with open source software, which is also why it is so important to involve experienced validation resources in the planning.
21.5 Risk management and open source software
As with any software application, in order to be cost-effective the validation of open source software should take a risk-based approach. This should follow established industry guidance such as ICH Q9, ISO 14971 or specifically the risk management approach described in appendix M3 of the GAMP® Guide.
An initial risk assessment should be conducted, as outlined in Figure 21.3, to facilitate the validation planning as described above. This should determine the overall risk severity of the open source software (with a focus on risks to patient safety, product quality and data integrity) and should consider the risks that result from both the package and supplier assessment.
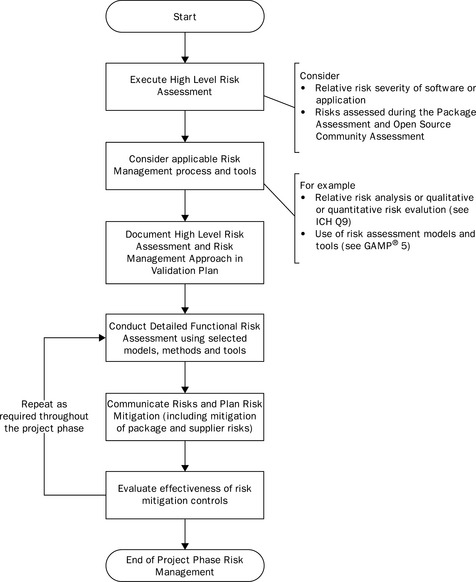
As part of the implementation or adoption of open source software, a detailed functional risk assessment should be conducted as described in the GAMP® Guide. This is the same process as for any other software package and should focus on the risk severity of specific software functions. This is in order to focus verification activities on those software functions that pose the highest risk.
However, with open source software, additional package and supplier risks may need to be considered as outlined above. Although the nature of the open source software will not change the risk severity, there may be specific issues that affect risk probability or risk detectability. These include:
It is likely that any increased risk probability and decreased risk detectability will increase the resulting risk priority, requiring additional scope and rigour with respect to verification activities (design reviews, testing, etc.). For each of these risks appropriate controls need to be identified, understanding that it may not be possible to leverage any supplier activities and that the regulated company will be responsible for any risk control activities.
Additional risk controls for open source software may include:
![]() more rigorous testing of standard software functions, where no formal supplier testing can be leveraged;
more rigorous testing of standard software functions, where no formal supplier testing can be leveraged;
![]() development of additional documentation, including user manuals, functional and technical specifications;
development of additional documentation, including user manuals, functional and technical specifications;
![]() development of rigorous change control and configuration management processes including detailed software release processes;
development of rigorous change control and configuration management processes including detailed software release processes;
The effectiveness of these risk controls should be monitored and reviewed to ensure that any risks are mitigated.
21.6 Key validation activities
Whatever the nature (categorisation) of the software, it is important that the regulated user defines and most probably documents clear requirements. Where there is little or no ability to change the functionality, it is acceptable for regulated company users to assess the package and confirm that it meets their requirements (Eudralex Volume 4 Annex 11). Even this will require a clear understanding of the business requirements, even if these are not documented in extensive user requirement specifications. It is recommended that the business process and data flows are documented as a minimum, against which the package can be assessed and this can be done at the package assessment stage.
For open source systems that are highly configurable, it is recommended that formal user requirements are documented and key areas are highlighted in Figure 21.4. It will be important to maintain these requirements in an up-to-date state as the use of the software changes over time. These may record user requirements in a variety of applicable formats including business process flows, use-cases, business rules and traditional singly stated user requirements (‘The system shall . . .’). It is usual to conduct a detailed functional risk assessment at this stage, examining each function (business process, use-case, etc.) to determine:
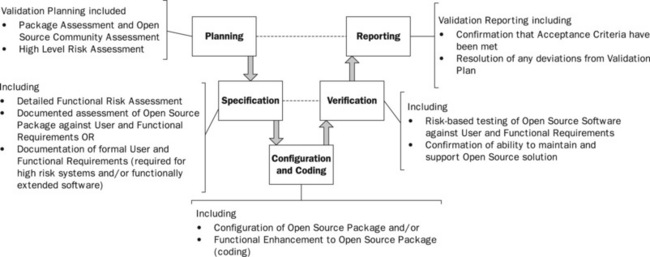
![]() whether or not the function requires formal validation, that is does it support regulatory significant requirements?
whether or not the function requires formal validation, that is does it support regulatory significant requirements?
![]() the risk severity of the specific function – usually assessed of a relative basis, but potentially using qualitative or quantitative risk assessment for software with a higher overall risk severity (see ICH Q9).
the risk severity of the specific function – usually assessed of a relative basis, but potentially using qualitative or quantitative risk assessment for software with a higher overall risk severity (see ICH Q9).
For functions that are determined to be regulatory significant and requiring validation, specifications will also need to be developed in order to document the setup (configuration) of the open source software by the specific regulated company. For COTS software this will include a record of the actual configuration settings (package configuration specification or application setup document). Where custom development is required this should also be documented in appropriate functional and technical design specifications.
It is worth noting here that where specific functions are developed by the open source community to meet the needs of a specific project, and where the community does not provide functional and technical design specifications, it is recommended that the regulated company’s involvement in the development includes the production of these documents. Not only will these provide the basis for better support and fault finding in the future, but they will also be of use to other life sciences companies wishing to use the same functional enhancements.
The nature of the open source movement implies a moral responsibility to share such developments with the wider community and it is common practice for developers in the open source community to ‘post’ copies of their software enhancements online for use by other users. While it is possible for life sciences companies to develop enhancements to open source software for their own sole benefit, this goes against the ethos of the movement and in some cases the open source licence agreement may require that such developments are made more widely available.
Where such functional enhancements require validation, the associated documentation (validation plans, requirements, specifications, test cases, etc.) is almost as useful as the software. Sharing such validation documentation (devoid of any company confidential or commercial details) fits well with the ethos of the open source movement and such documentation may be shared via open source forums or online repositories. Most open source software sites have a location where documentation can be shared and this practice is to be encouraged in line with the GAMP® key concept of leveraging ‘supplier’ documentation.
The open source community includes developers with widely varying experience. At one end there are professional software developers choosing to apply formal methods to the open source platform, and at the other are ‘amateurs’, developing software on a trial and error basis in their spare time, with little or no formal training and with little or no documentation.
Although all parties can make an active and valuable contribution to the development of open source software, there are inherent risks when regulatory significant applications are developed by those not formally trained in good software development practices. These risks may include:
![]() requirements are not fully understood, defined or documented, making it impossible to validate the software;
requirements are not fully understood, defined or documented, making it impossible to validate the software;
![]() the developed functionality may not fulfil the requirements due to poor specification and design;
the developed functionality may not fulfil the requirements due to poor specification and design;
![]() code may be excessively error-prone and inefficient;
code may be excessively error-prone and inefficient;
![]() important non-functional requirements may be missed, such as user authentication and secure password management, data security and data integrity;
important non-functional requirements may be missed, such as user authentication and secure password management, data security and data integrity;
![]() errors may not be identified due to insufficient or inappropriate testing.
errors may not be identified due to insufficient or inappropriate testing.
Actual examples of ‘bad practices’ encountered in open source software developed in this way include:
![]() failure to provide even basic user authentication in a web application accessing clinical trial data, allowing anyone with the URL to view personally identifiable health information;
failure to provide even basic user authentication in a web application accessing clinical trial data, allowing anyone with the URL to view personally identifiable health information;
![]() the storage of user IDs and passwords in an unprotected, unencrypted file;
the storage of user IDs and passwords in an unprotected, unencrypted file;
![]() the ability of a laboratory data capture routine to enter an infinite wait state with no timeout mechanism and no way of halting the program by normal means;
the ability of a laboratory data capture routine to enter an infinite wait state with no timeout mechanism and no way of halting the program by normal means;
![]() the ability for any user to access and change analytical data processed by an open source bioinformatics toolkit;
the ability for any user to access and change analytical data processed by an open source bioinformatics toolkit;
![]() complete lack of error handling at the applications level in a statistical program, allowing a ‘divide by zero’ error to halt the program with no indication of the problem.
complete lack of error handling at the applications level in a statistical program, allowing a ‘divide by zero’ error to halt the program with no indication of the problem.
Although it is possible that part-time, untrained developers can develop software that can be validated, this requires an understanding of both good software development practices and an understanding of software validation in the life sciences industry. Experience is that the development and validation of regulatory significant applications is best accomplished by real subject matter experts.
Where business process owners wish to contribute to the development of open source solutions, this is perhaps best achieved by defining clear requirements and executing user acceptance testing. In many cases the setup and use of the software may follow a ‘trial and error’ process – this may often be the case where documentation is poor or out of date. It is acceptable to follow such a prototyping process, but all requirements and specifications should be finalised and approved before moving into formal testing of the software.
Prior to formal testing, the software and associated documentation should be placed under change control, document and configuration management. It is unlikely that open source community controls will be adequate to provide the level of control required in the regulated life sciences industry (see below) and the regulated company will usually need to take responsibility for this.
It is worth noting that there are a small number of open source software tools on the market providing basic change control, configuration management, document management and test management functionality (e.g. Open Technology Real Services [24], and Project Open OSS [25] IT Service Management suites). Although these do not require formal validation, they should be assessed for suitability to ensure that they function as required, are secure and will be supported by the development community for the foreseeable future. Important areas to consider here are described in Figure 21.5.

Depending on the outcome of the risk assessment and the nature of the software, testing may consist of:
![]() unit and integration testing of any custom developed functions. This may take several forms including:
unit and integration testing of any custom developed functions. This may take several forms including:
o. white box testing (suitable where the custom function has been developed by the regulated company and the structure of the software is known),
o. black box testing (where the custom function has been developed in the open source community and the structure of the software is not known – only input conditions and outputs can be tested),
o. positive case testing, and where indicated as applicable by risk-assessment, negative case, challenge or stress testing;
![]() performance testing, where there are concerns about the performance or scalability of the software;
performance testing, where there are concerns about the performance or scalability of the software;
![]() functional testing, including:
functional testing, including:
o. appropriate testing of all requirements, usually by the development team (which may be an internal IT group or a third party),
o. user acceptance testing by the process owners (users), against their documented requirements (may not be required where a previous assessment has been made of a standard package that has not been altered in any way).
The use of open source software does not infer that there is a need to test to a lower standard. If anything, some of the additional risks arising from using open source software means that testing by the regulated company will be more extensive than in cases where testing from a commercial vendor (confirmed by a traditional supplier audit) can be relied on. All testing should follow accepted good practices in the life sciences industry, best covered in the GAMP® ‘Testing of GxP Systems’ Good Practice Guide. In some cases requirements cannot be verified by formal testing and other forms of verification should be leveraged as appropriate (i.e. design review, source code review, visual inspection, etc.). Supporting IT infrastructure (servers, operating systems, patches, database, storage, network components, etc.) should be qualified in accordance with the regulated company’s usual infrastructure qualification processes, and as required by Eudralex Volume 4, Annex 11.
All of the above should be defined in a suitable validation plan and reported in a corresponding validation report. Based on accepted principles, any of the above documentation may be combined into fewer documents for small or simple software applications, or alternative, equivalent documentation developed according to the applicable open source documentation conventions. Where the software is used within the European Union or used in a facility that complies with EU regulations – and soon to be any country that is a member of the Pharmaceutical Inspection Cooperation Scheme (PIC/S) – the documentation should also include a clear System Description (Eudralex Volume 4 Annex 11). The use of the open source software as a validated application also needs to be recorded in the regulated company’s inventory of software applications (Eudralex Volume 4, Annex 11).
In summary, the validation of open source software should follow a scalable, risk-based approach, just as any commercial software package. The only difference is that the inability to rely on a commercial vendor means that the regulated company may need to validate with broader scope and greater rigour than would otherwise be the case. This is likely to increase the cost of validation, which will offset some of the cost savings inherent in the use of open source software. This is why this needs to be assessed after the package and ‘supplier’ assessment – to confirm that the financial advantages for using open source software are still valid, despite the cost of validation.
21.7 Ongoing validation and compliance
Once the software has been initially validated, it is essential that it is maintained in a validated state and subject to periodic review and, in the case of any changes, appropriate re-validation (which may include regression testing of unchanged functions). This follows well-defined principles and processes best defined in the GAMP® Good Practice Guide ‘A Risk-Based Approach to Operation of GxP Computerized Systems’. Once again, the use of open source software does not provide a rationale for not following the processes defined in this guidance document, which are:
It is, of course, acceptable to scale these processes based on the risk severity, size and complexity of the open source software.
Key to maintaining the validated state is the use of an effective change control and configuration management process. This is often seen as contrary to the ethos of some parts of the open source community, where it is possible (and sometimes encouraged) for users to download and install the latest functional enhancements, patches and bug fixes – this speed of enhancement and the ability to leverage the efforts of other developers is undoubtedly one of the attractions of using open source software, especially in innovative applications. However, if this is allowed to happen the validated state of the software is immediately placed at risk and appropriate controls must be established. It is possible to allow this flexibility, but there are additional costs involved.
This requires a controlled set of environments to be established and these are outlined in Figure 21.6.
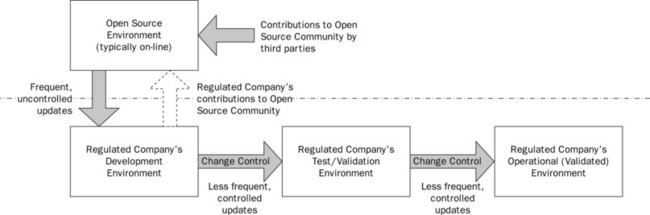
![]() A development environment for use by users and developers who wish to try out new functions, or even play a more participatory role in the open source community. This can be used to try out new functions available in the open source community or to develop new functionality, which may in turn be shared with the wider community.
A development environment for use by users and developers who wish to try out new functions, or even play a more participatory role in the open source community. This can be used to try out new functions available in the open source community or to develop new functionality, which may in turn be shared with the wider community.
![]() A test environment where new functions are tested – and old functions regression tested – prior to release into the production environment. This environment should be under formal change control, should be qualified in accordance with industry good practice and may be maintained and supported using formal processes and procedures.
A test environment where new functions are tested – and old functions regression tested – prior to release into the production environment. This environment should be under formal change control, should be qualified in accordance with industry good practice and may be maintained and supported using formal processes and procedures.
![]() An operational environment – which is under formal change control and subject to clearly defined release management processes. This environment also should be under formal change control, should be qualified in accordance with industry good practice and should be maintained and supported using formal processes and procedures.
An operational environment – which is under formal change control and subject to clearly defined release management processes. This environment also should be under formal change control, should be qualified in accordance with industry good practice and should be maintained and supported using formal processes and procedures.
Although it is possible for very frequent updates to be made to the development environment (including the download of new functions, bug fixes and patches) – sometimes weekly or daily – the release of such software to the test environment should have a clear scope, defined in terms of which updated requirements will be fulfilled. Release from the development environment to the test environment should be subject to defined version management and release management processes and will occur with a much lower frequency (every two weeks, every month or every three months depending on how much the open source software is changing).
The test environment should be formally qualified and subject to change control. This is to ensure that the test environment is functionally equivalent to the production environment and that testing in the test environment is representative of the production environment. Release of tested software should also be subject to defined version, release and change management processes and will again be on a less frequent basis. Although it is possible to ‘fast track’ new functions, the need to regression test existing, unchanged functionality places limits on exactly how fast this can be achieved. The production instance should, of course, also be qualified.
21.8 Conclusions
The validation process and controls described above apply only to validated open source software – those supporting regulatory significant processes or managing regulatory significant data or records. Non-validated open source software does not always need such control, although the prudent mitigation of business risks suggests that appropriate processes and controls should nevertheless be established.
The use of open source software does provide a number of advantages to life sciences companies and these include speed to implement new solutions, the ability to deploy ground-breaking software and the opportunity to participate in and benefit from a broad collaborative community interested in solving similar problems. However, the need to assure regulatory compliance and the requirement to validate and control such software does bring additional costs and add time to the process. Although this should not completely negate the advantages of open source software, the cost- and time-saving benefits may not be as great as in other non-regulated industries. Life sciences companies should therefore ensure that initiatives to leverage open source software are properly defined and managed, and that the relevant IT and QA groups are involved at an early stage. This will allow relevant package and supplier assessments to be conducted and the true costs and return on investment to be estimated.
Although much of the validation process is similar to the process used to validate commercial software, the nature of open source software means that there are some important differences, and the use of experienced subject matter experts to support the validation planning is highly recommended. Effective controls can be established to maintain the validated state and this will again require a common understanding of processes and close cooperation between the process owners (users), system owners (IT) and QA stakeholders.
In conclusion, there is no reason why free/open source software cannot be validated and used in support of regulatory significant business operations – it just may not be as simple, quick and inexpensive as it first appears.
21.9 References
[1] See http://www.simulations-plus.com/
[2] See http://www.opencrx.org/.
[3] See http://www.linkedin.com/.
[4] US Code of Federal Regulations, Title 21 Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals’.
[5] US Code of Federal Regulations, Title 21 Part 820 ‘Quality, System Regulation’ (Medical Devices).
[6] European Union Eudralex Vol 4 (Good manufacturing practice (GMP) Guidelines), Annex 11 (Computerised Systems).
[7] Pharmaceutical Inspection Co-operation Scheme PI-011 ‘Good Practices For Computerised Systems In Regulated GxP Environments’ (See http://www.picscheme.org/).
[8] Ref ISO 13485:2003 ‘Medical devices — Quality management systems — Requirements for regulatory purposes’ (see http://www.iso.org/).
[9] , US FDA ‘General Principles of Software Validation, 11 January 2002. see http://www.fda.gov
[10] See US FDA website at http://www.fda.gov/ for examples of Consent Decrees.
[11] , GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems, February 2008. published by ISPE – see http://www.ispe.org
[12] See http://www.knime.org/.
[13] See http://jas.freehep.org/jas3/.
[14] See http://rdkit.org/.
[15] See http://www.bioconductor.org/.
[16] See http://www.bioperl.org/.
[17] See http://biojava.org/.
[18] See http://bioclipse.net/.
[19] See http://emboss.sourceforge.net/.
[20] See http://www.taverna.org.uk/.
[21] See http://ugene.unipro.ru/.
[22] The R Foundation for Statistical Computing – see http://www.r-project.orgfoundation/.
[23] US Code of Federal Regulations, Title 21 Part 11 ‘Electronic Records and Signatures’.
[24] See http://otrs.org/.
[25] See http://www.project-open.com/en/solutions/itsm/index.html.