
Chapter 6
The Concentration Cell
Perhaps one of the more captivating and promising approaches to the practical storage of energy is in making use of the simple particles or billiard ball properties of matter. This view of the problem is enticing perhaps because of its direct simplicity. In other words, we might be able to make use of the straightforward “mechanical” properties of matter as described in classical molecular theory. In doing so, it appears we might be able to avoid many of the pitfalls of other energy systems that depend on specific properties of dissimilar materials (molecules), in which there are inherent mechanisms of degradation due to such issues as irreversible chemical changes, molecular diffusion from one region of the system to another, resulting in process contamination, or even just changes in molecular and physical structure that produce operational incompatibilities.
This approach does seem to offer ways around such life-limiting circumstances. In general, there is only one chemical species that participates in the energy storing process, and the processes all appear to be completely reversible by means of electrical input of appropriate polarity. If one is willing to design around or endure the peculiar electrical characteristics of such devices, perhaps a series of practical, low-cost systems can be produced in order to solve some of the more pressing energy problems we face in daily life.
We will proceed to outline some of the background physics and review the general behavior of concentration cells as electrochemical elements.
6.1 Colligative Properties of Matter
The various properties of a large aggregate of material particles that seem to behave as though they possessed properties that can be described as due only to their common “physical” or “mechanical” attributes are frequently referred to as colligative properties.
Webster’s Unabridged Dictionary defines colligative as “depending on the number of molecules or atoms rather than on their nature.” This more than implies that this class of attributes is independent of any chemical differences and is only concerned with the net effect of characterless particles regarding bulk behavior.
Examples of such properties and their effects include the following:
- Lowering of vapor pressure of a liquid
- Lowering of the freezing temperature of a mixture
- Boiling point elevation
- Osmotic pressure of solutions
- Electric potential difference between same ionic solutions with different concentrations.
At low concentrations of solutes in any of the above instances, the properties of the solutions are changed in a manner directly proportional to the amount of solute particles. This is especially true when their concentrations are so low that the properties of the solvent are unaltered and the interactions between solute particles are minimum. The vapor pressure depression, po – p, of a solution where po is the vapor pressure of the pure solvent, and p is the vapor pressure of the solution is expressible as
(6.1)

Xa is the mole fraction of the solute, a, in question.
Note that nowhere in these types of expressions does any property specific to the materials appear other than the vapor pressure of the solvent. However, we are concerned in these discussions only with the differences, po – p, that result from the introduction of any solute.
The idea of basing an energy system on a set of colligative properties is attractive because it might become possible to make a device that is independent of specific materials and, thus, symmetrical in construction, other than such factors as pressure, concentration, and temperature differentials between the same components of the device. Hence, we might be able to avoid any degradation in performance or life of that device due to the transfer of materials that differ in composition.
6.2 Electrochemical Application of Colligative Properties
The following is an initial technical description of the principles of operation of such a concentration cell. We will also refer here to the concentration cell as a Common Ion Redox cell (CIR), as a convenient abbreviation for this class of energy storage systems. These cells are, at times, also referred to as symmetrical cells since the cells are constructed the same at each electrode side. Electrodes are physically identical, electronically conductive, and chemically inert.
It makes use of the colligative properties of matter, or in this case, the large collections of iron atoms and ions in solution. In essence, the CIR cell employs the non-substance, specific properties of components and their physical behavior in an operating system. The raising and lowering of a solvent boiling point or freezing point as a consequence of the concentration of dissolved solute are colligative properties because they are virtually independent of materials composition. They are mostly strictly a result of the amount of solute molecules present in the liquid.
Another example of colligative properties is the interdependence of temperature and pressure of a gas (in the ideal gas range). These parameters are independent of the specific gas that is present and only dependent on the number of molecules present.
The search for a process, other than mechanical devices or electrochemical couples, that would enable the storage of energy in an inexpensive and reliable manner and that has a minimum of inherent failure mechanisms has resulted in the exploration of the characteristics of concentration cells as potentially practical devices for these applications. For the sake of brevity, we will refer to such cells as CIR devices (Common Ion Redox).
These devices are all redox in nature because, as is necessarily the case in chemical transformations, reduction and oxidation of chemical species occur within the device for the transference of energy. However, the label should not imply that these devices or cells are full flow designs with provision for removing or replacing electrolytes. They may be operated as either stationary, or static, electrolyte systems. The particulars of design and physical configuration depend on the intended applications. For example, it may be desirable to employ external liquid reservoirs, pumps, etc., in a full flow system if exceedingly long charge retention times are required. In such cases, the additional system’s complexity and costs may be justified.
This approach very closely resembles the process of the isothermal compression of a gas to store energy for later use. However, the energy input and output in gas compression is mechanical. A pump is employed to compress the gas, such as air within a confined volume, while dissipating the heat generated to the outside world. The input is usually a mechanical piston pump. To regain the stored energy in the form of a gas at higher than ambient pressure, the gas is permitted to expand back to the outside usually via a piston or centrifugal pump, thus returning some of that energy in useful mechanical form. It is also necessary to provide a heat exchange system to not only dissipate the thermal energy to the outside during compression but to also return thermal energy to the expanding and cooling gas.
In the CIR system, we are interested in a device that will operate with energy input and output directly in electrical form. In order to accomplish this end, it is obviously necessary to have electrical potential difference present, rather than a mechanical pressure, and charge transfers taking place within the device so that an electric current can flow in an associated external circuit. That circuit would provide the energy output as a current flow through an external electric load.
The energy potential difference between electrodes in a concentration cell is a consequence of the electrodes being immersed in electrolytes with different concentrations of the same chemical species. If the chemical species at these electrodes exist at different oxidation states, this energy difference is manifested as a net electric voltage between the electrodes. The electric field is proportional to a function of species concentration ratios.
This approach to storing energy is interesting because it appears that it is not only possible but also practical to obtain any level of electric potential in a CIR cell quite independently of the nature of the chemical agent(s), other than their solubility, electrical conductivity, and other general physical properties. Plus, the charge density is limited only by the amount of the chemical species that one can contain or compress in the locality of the electrodes.
The CIR cell is not limited to the electrochemical potential difference between two chemical agents. Instead, the voltage range is a continuum limited only by the ratio of concentrations that can be achieved by physical and mechanical methods. Some cell design possibilities are suggested.
Probably the most expeditious method of introducing and explaining the basic attributes and design parameters of concentration cells is to present this additional tutorial on the subject.
6.2.1 Compressed Gas
There are just a few steps in the explanation that will briefly describe some of the important, underlying fundamental principles of physics. Let us take a look at the ideal gas law,
(6.2)
where P = gas pressure, V = volume occupied by the gas, n = number of moles (number of molecules in that volume), R = universal gas constant, and T = temperature in absolute degrees.
Figure 6.1 shows a container with two compartments (equal volumes). These separate compartments can be connected by a gas pump to extract gas from one side to the other and by a valve permitting the compressed gas to flow (expand) from the high-pressure side to the other. The number of molecules initially in each side is n. If all the molecules were to be compressed into one side of the container, the compressed gas side would be at a pressure P′ = 2P. The amount of energy stored (isothermally) is then Liquid pump simply PV. Mechanical energy can be expressed as force times distance, or
(6.3)

Figure 6.1 Closed system gas compression.
Such a method for storing energy in this configuration is quite ineffective in terms of volume of space. Hence, the gas (air in this case) is usually compressed from the outside infinite supply source into a confined space such as the volume, V, of one compartment. Then the limitation of how much energy can be stored in that volume, V, is limited only by the strength of the container to withstand high pressure differentials between the outside (one atmosphere pressure) and its interior.
Not shown or discussed here is the considerable amount of temperature control and heat exchangers needed to maintain a near isothermal process. During the compression portion of a cycle heat, energy must be dissipated to the outside of the system, and during the expansion phase, heat must be supplied to the expanding gas.
6.2.2 Osmosis
Now let us turn our attention to another complementary process, or mechanism, known as osmosis. We can now move to a different environment, from that of a free gas to that of matter in the liquid state (much closer molecular proximity).
Figure 6.2 shows a simple diagram of a container with two compartments. A semi-permeable membrane separating the two compartments will more readily permit solvent (water) than the solute (salt, or other dissolved materials) to migrate through. This results in the “cell” developing an osmotic pressure differential, π, across that membrane. The value of π is analogous to the gas pressure in the previous relationship and found as

Figure 6.2 Open-air osmosis compression system.
(6.4)
In this case, v is the volume of the solution. In operation the solutes remain essentially in place, and the solvent, pressured by osmosis, moves through the barrier from the dilute side to the concentrated side until the hydrostatic pressure (or some externally supplied pressure) is equalized.
This is a reversible process. An externally applied pressure will move solvent through the semi-permeable separator, creating an increasingly dilute situation on the low-pressure side. The energy that went into creating a concentration differential can be reclaimed in part by permitting the osmotic pressure to reverse the situation.
Unfortunately, this process is not as practical for energy storage as the compression of a gas. In both instances, the process is mechanical and would require additional processes for the transformation into useful forms of energy.
6.2.3 Electrostatic Capacitor
An example of energy transfer and storage is that which is associated with the accumulation of electrical charge in a capacitor (condenser) as the result of an externally impressed electrical potential. Figure 6.3 shows such a simple arrangement in the form of a parallel plate condenser. This is a practical and widely used method for storing energy for brief periods of time in electrical circuitry, for load smoothing, and for providing large amounts of power for brief periods.
The relationship between charge and voltage is
(6.5)

Figure 6.3 Electrostatic capacitor.
where Q = electric charge in coulombs, V = electric potential in volts, and C = capacitance in farads.
A simple parallel plate capacitor has a capacitance, C, that can be represented as
(6.6)

where A = plate area, D = separation distance of plates, and ε = permittivity of the dielectric medium between the plates.
When the plates are connected to an electric potential source, charges flow from that source to the plates until the voltage across the plates, as dictated by the above relationship, just equals the source potential. The energy stored is
(6.7)

There is no “real” current flowing internally between the plates. However, a displacement current concept is employed to maintain continuity in the sense of a complete electric circuit. This is, again, an example of a concentration process to store energy. In this instance, the energy storing material in motion is not molecular but electric charges.
Capacitors provide a very useful and convenient method of storing for later use in large power applications.
6.2.4 Concentration Cells: CIR (Common Ion Redox)
Another approach to storing energy in direct electrical form, much as is accomplished in electrochemical cells that employ couplepotentials between dissimilar materials, is the electric potential obtained via concentration differentials with the same materials. Voltages can be produced within an electrochemical cell at the surfaces of electrodes if the concentrations of the same ionic species are different. This is expressed by the well-known Nernst equation:
(6.8)

where z = electric charge of the specific ions, F = faraday number, a1 = activities at electrode (1), and a2 = activities at electrode (2).
A more convenient and a more easily calculated value for the concentration voltage is the substitution of reagent concentrations for their activities. This practice results in calculated values of electric potentials that are not as accurate as would be obtained if activities were employed but are reasonably valid if the concentrations are low. However, the ratios of the activity coefficients at different concentrations of specific ionic species do not differ much over a wide range on concentrations. For our purposes here, the agreement is close enough to provide a working basis for preliminary estimates of cell potentials. The table below shows fairly constant values for activity coefficients over a wide concentration range. This is true for weak as well as strong electrolytes.
The activity, a, of an ion or ionic compound is related to the activity coefficient, γ, by the simple equation
(6.9)

where c is the concentration in molarity, as listed in Table 6.1, multiplied.
Table 6.1 Activity coefficients, γ, at 25°C.
| Molarity | HCl | NaCl | NaOH | CaCl2 |
| 0.005 | 0.930 | 0.928 | …… | 0.789 |
| 0.01 | 0.906 | 0.903 | 0.89 | 0.732 |
| 0.05 | 0.833 | 0.821 | 0.80 | 0.584 |
| 0.10 | 0.798 | 0.778 | 0.75 | 0.524 |
| 0.50 | 0.769 | 0.68 | 0.68 | 0.510 |
| 1.00 | 0.811 | 0.656 | 0.66 | 0.725 |
| 2.00 | 1.011 | 0.670 | 0.68 | 1.554 |
From: Physical chemistry by Alberty & Daniels, 1955, John Wiley & Sons.
The activities for HCl at 0.005 molarity and at 2.00 molarity multiplied by their respective concentrations are a = 0.930 × 0.005 = 0.0047 at c = 0.005 and a = 1.011 × 2.0 = 2.022 at c = 2, which are vast differences for a.
Figure 6.4 is a simple representation of such a cell where the two electrode compartments are separated by a microporous or ion transfer membrane. The specific species is relatively unimportant, except for its solubility, mobility, and concentration as seen by each electrode.

Figure 6.4 Simple electrochemical cell.
If the chemical species were merely a compound such as NaCl that ionizes in the solvent (water in this example), there would be no electric current flow possible even if the concentrations at the two depicted electrodes were vastly different. Only an osmotic pressure difference would be realized and no electrical potential.
In order to have ionic substances migrate from one electrode to another within the cell, it would necessitate an accumulation of opposite electric charge at the electrodes (as occurs in a capacitor), and there must be a closed circuit through a means external to the cell to provide an external electric current flow. The former condition is not possible because the electrolyte between the electrodes is conductive. Hence, no static electric charge can be sustained, and the latter can occur only if there is an electric potential across the cell.
In order for an external current flow to be developed, there must be some mechanism at the electrodes to take on and give up electric charges (electrons). A process that can be used to complete the charge exchange cycle at electrodes is that of moving up or down the scale of state of oxidation or reduction for an element or compound. Then, a continuous path for electric charge flow within the electrolytic cell as well as electron migration through the external circuit can be provided.
It can readily be seen that we now have a compression procedure (concentration differential of the same molecular species) that lends itself to a reversible process for the storage of energy in electrical form. The general exchange process below illustrates this for a species that we will identify as A, with two oxidation states, A+n and A+(n–1). Then, ![]() .
.
As a very simple situation to illustrate the principle, let us assume that the element or species exists in solution as a compound, AxBy, with another species, B, that does not change oxidation state. Then, the process, as shown in Figure 6.5, would be the transport A ions across a separator from one side to the other. Furthermore, electrons would flow in the external circuit to maintain total electrical neutrality.

Figure 6.5 Charges stored by surface adsorption.
Examining the situation quantitatively, we see that, in a cell as described in Figure 6.4, the maximum concentration of solute is limited by the solubility of that compound. In most cases, the solubility of such ionic materials is in the range of 3 to 6 molar. If A carries a single transfer charge per ion, then the maximum charge density for a given volume of 4 molar solution would be 2 × 25 AH per mole, or about 100 AH per liter per side. That figure reduces to 50 AH per liter of total volume of cell. Even with the 4 molar limitation and only one charge carrier per ion, the charge density is still quite attractive.
However, one must look at the voltage at which the charges are delivered to an external load. For a simple cell in Figure 6.4, the voltage rises as the cell is “charged,” meaning that the concentration of ion A increases and proportionately decreases on the respective cell side. Total cell volume is 1 liter, as shown in Table 6.2 below.
Table 6.2 Calculated voltages vs amp-hour input.
| C1 | C2 | AH input | End voltage | Energy stored |
| 3.9 M | 0.1 M | 50 | 0.05 | ~1/2(2.5) Wh |
| 3.99 | 0.01 | 5 | 0.10 | ~1/2(0.5) |
| 3.999 | 0.001 | 0.5 | 0.15 | ~1/2(0.075) |
As one can see, the energy stored and the available voltage both rapidly diminish as the cell bulk concentration difference progressively diminishes.
The storage of reagents at the electrode sites in extremely concentrated form is one of the numerous contributions or innovations. Figure 6.5 diagrammatically illustrates this effect. The reagents are stored as uncharged molecular structures within sites by “ion injection” and van der Waals-type forces. These molecular species are slowly released as ions in solution in the immediate vicinity of the electrode surfaces in accordance with the demand during discharge.
The important issue in assessing or computing the performance of cells and their operating voltages is to realize that the cell potentials are the result of the electrolyte environment in the immediate region of the electrode surfaces (frequently referred to as the Helmholtz layer). Conditions in the bulk electrolyte region are mostly irrelevant with regard to electrode potentials. Only what the electrodes “see” to some limited distance away is important.
To use the example above of active species A, the potential of a concentration cell is expressed essentially as
(6.10)

Cells at TRL have been operated with as much as 1.2 to 1.4 volts open circuit. Such high voltages would entail concentration ratios of between 1020 and 1024 for the logarithm to the base ten to be high enough to achieve these potentials.
It may seem that somehow the cell operation violates some fundamental law involving conservation of energy or a related issue. Even though the cells depicted in Figures 6.4 and 6.5 are almost identical, because their volumes are the same and the reagents are the same due to the electrode structure and mechanism of storage, one gives immensely greater voltages. Note that the charging takes place at much higher voltages. Hence, the energy input, or work done on the charged ions in “compressing” them into storage at high molecular densities, is also great. As in compressing a metallic spring, the force needed to further compress the metal coil will increase as compression progresses, but that energy is available upon “discharge,” minus some irreversible frictional heat losses.
This CIR process may be regarded as analogous to a molecular (ionic) spring. The important issues regarding the CIR approach are its very low cost, extremely long life, and abuse resistance. These are the prime factors that motivated exploring the concentration cell approach. In addition, it is an approach that departs in principle from all other batteries. This system depends solely on concentration differentials of the same materials. Hence, the problems of cell materials and structure corruption due to irreversible diffusion and non-uniform deposition of active reagents are absent.
This approach to energy storage enables us to avoid irreversible materials transfer problems and presents an opportunity for exploring many different materials combinations as well as the relatively unlimited possibility of making cells with very high voltages.
A brief comparison between the full flow and a static electrolyte does point out the obviously greater complexity of the former. Problems with full flow include the following:
- Maintaining reasonably uniform flow through many parallel cell channels
- Additional hardware of tanks and plumbing, pumps, and manifolds
- Increased complexity of battery module with feeder tubes, etc.
- Additional power required for pumps
- Self discharge losses when sitting idle with flow
- Necessity to build hardware as pressure vessels
- The static cell method avoids all of these problems.
6.3 Further Discussions on Fundamental Issues
The following is intended as a quick review of the origin of concentration potentials in an electrochemical cell. Sometimes these potentials are referred to as polarization voltages resulting from starvation at electrode surfaces of reactant containing electrolyte. Concentration cell behavior will be developed here in terms of familiar chemical principles.
First, we must define the meaning of half-cell potential to understand the development of the concepts of concentration dependent voltages.
The kinetics, diffusion equilibrium, sorption rates, and electric field penetration depths are not discussed at this time because they don’t contribute to the basic understanding. There are trade-offs between storage capacity and discharging ability. However, none of these issues are necessary in order to comprehend the technical approach and its configuration.
The main purpose is the identification of the important issues in a step-by-step fashion so as to grasp the physical essentials of a concentration cell.
Let us examine the potential between a metallic electrode and its ions in solution in its immediate vicinity. Consider the familiar configuration of a copper plate immersed in a copper sulfate solution. One may reasonably ask what the electric potential is between the copper and the solution of its own ions, as shown in Figure 6.6. Unfortunately, there is no physical way of making such a measurement without the presence of a second electrode. Such measurements have long ago been standardized by the use of either a silver/silver chloride, a calomel electrode, or a hydrogen reference electrode.

Figure 6.6 Standard reference electrode potentials.
In general, whenever electric potentials of the various elements are given, they refer to a hydrogen electrode, and that hydrogen reference is arbitrarily set at zero. In this case, the cupric potential is +0.337 volts with respect to hydrogen:
(6.11)

Since the potential of any electrode in an electrolyte is dependent on the concentration of the specific ion involved in the attendant reaction, we know that the voltage for copper is determined at a standardized electrolyte (copper ion) concentration. In most cases, this has been standardized as a concentration in which the “activity” is unity. Usually, the condition for activity = 1 at STP is in a 1 molar solution. The standard hydrogen potential is established at 1 atmosphere pressure of H2 gas phase and surrounded by hydrogen ions at unit activity.
The above is important because one must establish a series of reference or standardized conditions in order to explore any of the cell properties. In the classic Daniel Cell, we have the two metals Zn and Cu in solutions of their respective salts. In Figure 6.7, a porous barrier separates the solutions. The potential between the two electrodes is as follows:
(6.12)


Figure 6.7 The Daniel cell.
Now, let us look more closely at the situation encountered by an electrode when it is immersed in electrolyte at a “bulk concentration” of any specific solute or ions. The diagram shown below in Figure 6.8 is a standard representation of an interpretation of ion concentration and electrode potential in view of distance from an electrode.

Figure 6.8 Electrolyte regions at electrode surfaces.
The above tells us clearly that there must be “intimate” contact between the electronic conducting electrodes and the molecular Helmholtz layer species undergoing electron exchange. This all happens within a very short distance. The usual porous electrodes are simply not useable here. Their site distances are too great. If we are not able to maintain the intimacy, then the cell will be very severely limited in performance by diffusion to the outer reaches of the bulk electrolyte.
An electrode immersed in a static electrolyte (non-flowing) sees only a small distance into the surroundings. This distance is not much more than into the beginning of the Gouy diffuse layer. The layer, referred to as the Helmholtz layer, is essentially an electric double layer. The voltage, ∆φ, between the electrode and the remainder, or bulk electrolyte, is given as
(6.13)

The distances involved are quite small. In most instances where electrolyte concentrations are appreciable, the diffuse layer is in the order of 10–7 cm, or a few ionic (molecular) diameters thick.
Obviously, if electric current is produced at the electrode surface, or if there is significant flow in the surface region, the potential diagram above would be distorted accordingly.
We can now return to the matter of electric potentials and concentration conditions. In fact, we will use one of the specific couples we have explored as a quick example.
Consider the cell (see Figure 6.9) between hydrogen and an inert electrode such as Pt or carbon. One side of the cell is a hydrogen probe in an acid solution (H+ ions), and the other is a carbon electrode immersed in a solution of a mixture of ferric and ferrous ions (perhaps ferric and ferrous chloride). The anion, chloride, has little to do with the whole process.
The cell reaction is
(6.14)
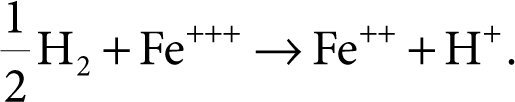

Figure 6.9 Hydrogen reference electrode cell.
Applying the Nernst equation, which says that the potential is proportional to the logarithm of the ratio of the activities, to determine voltage,
(6.15)

but since aH+ = 1 and aH2 = 1,
(6.16)

Eo is the electromotive force of an inert electrode surrounded by equal activities of ferrous and ferric ions when measured against the standard hydrogen electrode. When aFe++ and aFe+++ are equal, the last term becomes zero. These activities have direct relationships to concentrations, and in some instances (dilute solutions) it is possible to substitute concentrations for activities as a very first approximation of cell voltages.
Now imagine a cell wherein both electrodes are chemically inert (carbon) and are immersed in a mixed solution of ferrous and ferric salts. The net potential between them would obviously be zero, if for no other reason than the symmetry or sameness of both sides of a cell.
An oxidation/reduction process does take place but strictly to provide a mechanism for electric charge transport. In no way does this redox contribute to either the cell voltage or the energy level. As you can see from the basic thermodynamic equations resulting in the Nernst relationship, nowhere do the properties of the specific reactants appear other than their concentration.
Figure 6.10 illustrates a simple concentration cell for non-porous electrodes.

Figure 6.10 The iron concentration cell.
Both sides are the same except for the differences in concentrations of ferrous and ferric salts (not ions by themselves). Not shown is the fact that the solution is acidified and conduction is mainly by H+ ions. Cation membranes are employed, and iron is notoriously sluggish as a charge carrier. All reagents, whether they are ferrous, ferric, or sulfide salts in polysulfide cells, are assumed to be in uniform solution in the bulk electrolyte volume on their respective sides of a cell.
Cells with more practical characteristics do not have as simple a configuration as those shown above. In order to design them for more practical uses, they store reagents in a very concentrated (or very diluted) format within the electrode structures themselves. Such configurations are shown and described in considerable detail in the pages to follow.
6.4 Adsorption and Diffusion Rate Balance
The following is a description of the principal aspects of a concentration cell as configured in this development and is in the form of a device where the internal charges are ions in “solution.”
There are few chemical elements and compounds that lend themselves well to such processes. The active material chosen as representative of this class of device employs sulfur as both oxidizer and reducer.
In addition to treating the diffusion rates through a separator into and out of the bulk storage regions, the rates of adsorption/desorption must be taken into account. As a first approximation, let us use the expression by Langmuir regarding adsorption isotherms. This approximation does not account for changes in adsorptivity as the surface sites become more occupied, nor does it account for any changes in the ratio of the coefficients αa and αd, the adsorption and desorption, in the constant relationships below.
(6.17)

C is the concentration of the species in solution being adsorbed, or the adsorbent, and θ is the fraction of the total available sites that are occupied by adsorbent at point in time.
The introduction of this new term changes not only the mathematical balance equations but also the very nature of the mechanisms of storage. Now the electrode is no longer just seeing the concentration of specific ions in the surrounding bulk electrolyte, but it primarily sees the concentration of the adsorbed ions readily available at the electrode surface. Therefore, it becomes necessary to modify the model and our way of thinking about what may be happening within the cell.
The rates are as follows:
- Rg = generation rate of S= ions, always at the (–) electrode = KgI
- Rs = adsorption rate = α1(1–θ)C
- Rd = desorption rate = αdθ
- Rm = net diffusion rate across the membrane = Km/V(2Q1 – Qo).
In more general terms, this may be expressed as a sum of differentials as follows.
Figure 6.11 is a diagram of the “compartmentalized” nature of the cell, with the “Helmholtz” region being essentially what the electrode sees, and is the concentrated electrolyte in dynamic equilibrium with its solid forms on the surface of the porous electrodes. This region of electrolyte is also in dynamic equilibrium with the bulk electrolyte occupying the volume between the electrodes and the separator membrane. The bulk concentration differentials across the separator determine the diffusion rate of soluble components, e.g., sulfur complexed with sulfides and sulfide ions from one side of a cell to the opposite side.

Figure 6.11 Half-cell representation.
Low solubility versus high salt solubility is an interesting issue. Also, we want salts to go into solution fast to sustain higher cell currents but to also precipitate out of solution for higher retention purposes.

In equation (6.18) the terms are defined as follows:
- (dQ/dt)net is the net rate of increase of the specific ion.
- (dQ/dt)i is the rate of ion production by charging electric current.
- (dQ/dt)diff is the loss rate by diffusion away from the electrode. (dQ/dt)ad is the loss rate from solution by adsorption.
- (dQ/dt)ppt is the loss rate by precipitation of their compounds.
Thus, there are the balances between the rates of desorption as well as the solubilization of the precipitated reagent, in this case, Na2S.
The rates of sorption and diffusion are placed in the loss category since they represent losses from the (–) electrolyte. Furthermore, desorption and the electrical generation rates are to be considered gains, or sources of S= ions to the (–) electrolyte. In this fashion, we can handle the ensuing balance equations. Thus, the net rate, R, into the (–) electrolyte can also be represented by equation (6.19):

where Rg is the production rate by electric current, Rm is diffusion rate, Rs is the sorption rate, and Rd is the desorption rate.
More specifically, the above becomes
(6.20)

6.5 Storage by Adsorption and Solids Precipitation
The most important parameters for optimum cell operation are (1) to maximize energy capacity by increasing the amount of charge density per unit area of electrode and (2) to establish high and sustainable concentration ratios of ionic components, i.e., large concentration at the (–) electrode and small concentration at the (+) electrode.
One way to enhance charge capacity while reducing diffusion losses is to make use of solid precipitates. If the solubility of the sulfide compounds is exceeded, then they will precipitate out of solution and, by design, onto the surfaces of the electrodes. This provides for the additional supply of reagents and in a form that will remain within the (–) and (+) cell compartments for longer periods of time.
If we stipulate a simple linear relationship between the solution and dissolution (re-solubilizing) rates for the solid sulfur and polysulfides that fall in and out of solution, we can express this additional factor as follows.
Let the rates Rp1 and Rp2, the rates with which the compounds are precipitated and dissolved, be represented as
(6.21)

where β1 and β2 are constants at any given temperature, C1 is the concentration of the S= ions in solution, and P is a constant associated with the amount of solid Na2S in solid precipitate form. The term Cs is the maximum concentration that the electrolyte will tolerate prior to “salting out.” This last term is not a constant and tends to be very dependent on conditions such as temperature, presence of suspended solids, etc. The concentration tendencies of C1, being somewhat above Cs, will drive the precipitation of material out of solution. We can assume, for the sake of simplicity, that the relationships are linear.
Thus, the complete rate equation takes the form
(6.22)

The net quantity of interest to us at the end of charging is the amount of S= ions available for discharge. That is found by equating the input rates and loss rates at dynamic balance for any charging current, I, as the maximum achievable charge, which occurs when R = 0, or when
(6.23)

Our main interest in the above derivations is the evaluation of the amount of species, Qa, adsorbed within the electrode. In this case, it’s the sulfide ion in the form of the electrically neutral compound, sodium sulfide. (Ions cannot be adsorbed as such without the accumulation of an inordinately high electrical charge).
It is necessary then to put 0 into terms of quantity of material rather than the ratio of occupied sites to total available sites. This is easily accomplished.
If we let A = the total number of available sites (per unit electrode volume), then the factor (1–0) can be replaced in terms of A:
(6.24)

The only explanation for the magnitude of voltages obtained from our experimental cells is that the mechanism of “electroadsorption” or its equivalent takes place. That would necessitate a small layer of “stagnant” electrolyte at the electrode porous surfaces. This layer might be thought of as a very dense cloud layer, δ, of concentrated species (S= ions) that are about to be adsorbed.
The balanced rate equation is thus modified to reflect this micro-layer assumption as
(6.25)

where Cδ is the concentration of S= in the immediate neighborhood of the electrode and precipitate surfaces.
Substituting the expression for θ in terms of Qa, the amount adsorbed, we get
(6.26)

The time delay between adsorption and generation by electric current and charge transfer largely gives rise to this δ layer of not much more than a number of molecular diameters, or mean free paths in thickness.
It is very important to the successful operation of such concentration cells, regarding their practical application, that the capacity and charge retention are not entirely, or even largely, dependant on membrane characteristics. Otherwise, we would be engaged in the continuing compromises between electrical conductivity and diffusion coefficients of such materials. Virtually everything that is done to reduce separator electrical resistance also promotes molecular diffusion in membranes. Hence, we seek mechanisms wherein molecular species can be collected to very high concentrations by some sort of bonding or retardation process, while not significantly detracting from either the cell potential or ionic mobility. The membrane serves the purpose mainly of keeping the two bulk electrolytes apart. A high effective concentration of the ionic species of interest (the S= ion in this instance) must be established and maintained throughout the charging process in order to “force” the diffusion of that ion into the carbon surfaces to be adsorbed.
A gel electrolyte might very well serve that purpose. Some of our experimental results have shown that excellent operation can be obtained with only a gel electrolyte to immobilize the substances. However, it is necessary to pay attention to the mechanism of electrode starvation when employing gels because the S= ion can be depleted in the δ layer, resulting in high resistance and little charge transfer. The risk with gel electrolytes is that a large amount of the total reagents in the needed oxidation state can be trapped too far from the reaction sites for prolonged periods of time, becoming essentially unavailable for electron exchange.
6.6 Some Interesting Aspects of Concentration Cells
- All active materials employed have some solubility, enabling all solids that are formed during the cycling processes to be returned to new and uniform positions within the cell.
- The cell is symmetric in the materials sense, i.e., the active materials are the same throughout the cell. However, they are not symmetric with regard to oxidation state population densities.
- There are only two oxidation states available with materials selected, hence the symmetry at discharged condition.
- Ionic, energy-storing components are stored within electronically conductive, high surface area pores of electrodes, thus enabling high coulombic capacity.
- To further increase coulombic capacity, active materials are stored in quantities that exceed their solubility in the electrolytes. These precipitated components are deposited and stored also within the pore structure of electrodes so that they may be readily available for re-solubilization and subsequent participation in the electrolytic, energy producing reactions.
- To obtain and maintain high electric potentials, the concentrations of reducing and oxidizing agents are replenished by the respective precipitation of components. An example of this is during the discharge mode of a cell at the positive electrode. As the sodium ions arrive, the sulfur stored in that electrode solubilizes and generates sulfur ions in association with the newly arrived sodium ions, thus maintaining a high sulfide ion concentration within the close (Helmholtz) region to contribute to cell voltage. A similar, but opposite, process takes place at the negative electrode where solid sodium sulfide salt is precipitated out of solution.
- All active components are electronically non-conductive to prevent internal short circuit situations that are encountered with metal plating, dendrite growth, etc.
For the sake of simplicity, let us replace activities with concentrations R and L for the right and left sides, respectively. Now the expression for net cell potential, E, can be represented as
(6.27)

Examining this cell more closely will reveal some serious limitations regarding maximum attainable voltages and energy densities. Even if we had a perfect barrier (separator with zero resistance and zero unwanted diffusion or transport), the first limitation would be due to the maximum solubility of the iron salts. They are limited to about 3 molar at room temperature. That means that the limitation would be 1.5 molar solution on each side at the start (assuming equal volumes on either side). Looking now at the voltages that might be expected, we see that, if upon charging we went from 1.4 M to 2.9 M of ferrous on the left side, ferrous concentration would be down on the right side to 0.1 M, and, correspondingly, the ferric ion concentrations would be up on the left to 2.9 M and down to 0.1 M on the right. A total charge transfer could be calculated on the basis of cell volumes.
Substituting these values into the Nernst equation above, we get
(6.28)

Even if we were able to achieve concentrations of 0.01 M and 3 M on the respective sides, the maximum voltage realizable would be about 0.25 volts. Let’s now look at the total electric charge available. Taking the maximum molarity change of 1.5 and a volume of 1 cm2 per cell side, the maximum charge transfer would be amp-hours at an average voltage of 0.15 volts. Obviously, this needs to be improved. The greatest limitations to attaining higher voltages and energy densities are due to two principal factors:
- Limitations of solubility of reagents
- Subsequent limitations to concentration differentials
Hence, we cannot be limited by the solubility of reagents if we want high ED. We must, therefore, synthesize another means of keeping the reagents around and available. Reagents are, in this example of an iron cell, ferrous and ferric compounds. If we try to increase bulk electrolyte concentration, the reagents will merely precipitate out and fall to the bottom. However, an actual concentration cell would not have a bottom as such because very porous, conductive materials would occupy most of the intervening space in a cell. In order to overcome these factors, we may resort to storing reagents in an adsorbed or “pseudo-solid” form within electrode structures themselves. This single feature enables us to go well beyond solubility limitations as well as developing and maintaining concentration ratios for higher voltages. It is especially possible to achieve these ends because reagents in solid form are not metallic and do not conduct electronically. Figure 6.12 shows, in principle, this approach method.

Figure 6.12 Simple non-mobile electrolyte concentration cell.
The entire intervening space between electrodes and the separator is occupied by extremely high surface area and electronically conductive carbon. These micro or nano-porous particles are selected for their conductivity range, pore size, and pore distribution.
The carbon employed has an available surface area measured as between 1,500 to 2,500 m2 per gram. This corresponds to a structure whose walls are in the range of 2 to 6 atomic diameters thick.
Much of the reagents are stored in an interstitial state on the carbon structure. Estimates based on these kinds of data and experimental findings indicate it might be possible to attain performances equivalent to 10 to 15 molar solutions.
Also, by the self-constricted, controlled ionization constants within these structures, it appears that we are able to attain effective concentration ratios of 1010 to 1014 from each oxidation species. In looking at the basic equation for the voltage, it can be seen that, in order to achieve a cell potential of even 1.2 volts, the logarithm term in the expression for voltage must be as high as 20.
An additional benefit is the reduction of diffusion losses (increased charge retention) because much of the reagents are stored in an interstitial solid state. Such cells and their electrodes must be fabricated and prepared before assembly to utilize such compactness.
There is a balance between undissociated and ionized molecular ferrous and ferric compounds as part of the storage and transport release processes. Our understanding of this and various other processes and energy level change mechanisms is not yet clear, but progress is being made.
The attractiveness of these cells is their simplicity and their reversibility for an indefinite number of times.
6.7 Concentration Cell Storage Mechanisms that Employ Sulfur
In the CIR system it is necessary to store reagents in very concentrated form at the electrode surfaces themselves. The reagents can be stored as ions in the form of soluble compounds or as their solids ready to go into solution as needed during charging/discharging modes.
Bulk storage of reactants, oxidized and reduced state ions, in the electrolyte as dissolved compounds provides very low voltages and specific energy storage density. Most compounds of useable materials, such as those of iron, sulfur, bromine, copper, etc., are limited to about 4 molar at standard temperatures and pressures. Such limitations in solubility give rise to small energy densities.
The cells with which we are concerned here are classified as cells with liquid junctions and in which transference takes place. The sulfur system will be our first analytic model for determining the balance of materials at the beginning and end of a charge cycle. If we were to regard only the soluble forms of the sulfur salts, e.g., sodium, potassium, or lithium compounds, the maximum ED attainable is calculated as follows.
Let us assume as a first approximation that no polysulfides are formed and that the process is strictly between sodium monosulfide and elemental sulfur. Also, we need to start any charge cycle with the same materials with their concentrations equal on both sides of a cell.
The minimum concentrations for balance would be 2 Na2 // 2S (fully charged), where // indicates a divider or ionically conducting separator between cell compartments. Also, we will assume for simplicity that the two cell compartments are equal in volume and that only sodium ions are transported across the separator.
Let us take this step to discharge. The discharged situation would be Na2S + 2S // Na2S + S, which is a symmetrical balance with no difference in concentration on either side of Na2S or S. The net transfer of charge per liter per side would be 50 AH.
The next workable concentration for balance is an even number of moles, or 4Na2S // 4S fully charged, 2S + 2Na2S // 2Na2S + 2S discharged state.
The net transfer per liter per side here would be 100 AH. The next, of course, would be 6Na2S // 6S charged, 3Na2S + 3S // 3Na2S + 3S discharged.
Net charge transfer per 2 liters total of volume is 150 AH. In generalized form, if n is any integer, then the process proceeds as above as 2nNa2S + 2nS // 2nNa2S + 2nS charged, and the net charge transfer per total liter volume is simply n × 50 AH.
Let us examine briefly the structure of the final reactants. If the highest possible polymers were formed upon discharge, they would be the dimers, or Na2S2.
If the cell electrodes were unable to effectively (reversibly) store elemental sulfur, then we would need to resort to the maximum polysulfide at the charged state. That seems to be the pentasulfide, Na2S5. Then the cell at total charge might be xNa2S // yNa2S5, and thus limited in reactant weight by the necessity for higher ratio of sodium ions per sulfur atoms present.
Let us start with a one molar solution of the pentasulfide on one side of the cell and the appropriate molar monosulfide on the opposite side so that we conclude at the end of discharge with the same materials at the same concentrations on each side. The steps toward total discharge are as follows:
- XNa2S // Na2S5
- (X–1)Na2S // Na2S4 + Na2S
- (X–2)Na2S // Na2S3 + 2Na2S
- (X–3)Na2S // Na2S2 + 3Na2S
- (X–4)Na22S // 5Na2S
Hence, the value of X = 9 makes the balance exact at the discharge end. The total charge from this 9 molar mono-sulfide solution would be 200 AH, and the total weight exclusive of water would be 862 grams. The ED for the dry salts at 1 volt is then 105 WH/lb.
Now we shall examine the possible mechanisms for storing the materials in their two states of oxidation. The goal is to structure a system such that a maximum of concentration difference can be achieved between S–2 and S on the two opposite electrodes. It is important to sustain that high voltage over the longest portion of the discharge, or charge delivery.
6.8 Species Balance
There are three explanations possible that, at this point, could describe the situation of affairs within the cell at any state of charge. First, let us assume that all species remain solution at all times, that the only charge carrier within the cell is Na+ ions, and that there is no net migration of sulfur from one cell side into the other. Vertical bars, ||, enclose concentrations. The two sides of a cell (opposite the separator) are indicated by subscripts a and b:
(6.29)

and
(6.30)

The above also assumes that the sodium sulfide exits within the pores of the carbon electrodes as ionized molecules. This assumption seems not to satisfy the very high cell potentials experimentally measured in the laboratory with solute concentrations in the range of 2 to 3 molar. We will later examine the diffusion rates and possible high concentration differentials in such a configuration.
Secondly, it seems more likely, however, that the species sulfur, polysulfide or monosulfide, would partially be in either a solid state or in an adsorbed state on the pore surfaces. If so, the balance of materials may be expressed as
(6.31)

where the subscripts relate to the cell sides a and b, and i and s indicate ionic or solid state, respectively. The brackets contain the absolute quantities of the respective ions, or atoms, in either the a or b side of the cell.
There is also the possibility that these materials may exist not as solids but in the form of an “interfacial state.” See “Physical Chemistry” by E. A. Moelwyn-Hughes, Pergamon Press, 1957, pages 894–950.
6.9 Electrode Surface Potentials
It is important to consider whether the cell configuration is one with or without transport in order to account for any liquid junction potentials supplementing the electrode voltages.
Nernst’s relationship does not account for the very high electric potentials attained in laboratory test cells solely on the basis of measured bulk electrolyte concentration differences on either side of cell separators. Since there are no other significant reactions taking place the electrodes must be experiencing ionic concentrations ratios orders of magnitude greater than the bulk electrolyte ratios.
The search then begins for a plausible explanation of how a cell with no other energy related processes taking place, other than the difference in concentration of the two oxidation states of the same chemical element, can produce such high potentials and sustain high electrical charge densities. If there are no other processes involved, then we must look toward some mechanism whereby the chemical species in question are able to be accumulated at the electrode surfaces and give rise to such high voltages.
At the present time, it is speculated that the specific ionic species, in this case the ferric and ferrous ions, are injected by the process of electrosorption directly into the pore regions of the microporous carbon at very high effective concentrations. In the case of a concentration cell based on iron chemistry even though the maximum concentrations of the compounds of ferric and ferrous chlorides are limited to not much over 3 molar at room temperature, the population densities of the adsorbed and stored ferrous and ferric ions become equivalent to extremely high activities seen by the electrodes. In order to accomplish this, more energy than what is normally needed would be required to separate the oxidation states during the “charging” process. This condition, for the maintenance of conservation principles, has been observed during cell cycling in terms of the volt-amp inputs and outputs.
There are undoubtedly many processes and mechanisms that take place at the electrode surfaces that call for greater understanding and quantifying. Let us first list and then examine the physical and chemical activity possibilities. Omitting the chances that there are any net or permanent chemical changes occurring in the electrical cycling of the cell, the following are possible, reversible processes:
- Exceeding salt solubility at electrodes during charging, resulting in solid compounds at the surfaces with attendant free energy changes (energy of ionization and dissolution)
- The creation of immense concentration ratios of S= in the sulfur based cell and of Fe++ and Fe+++ ions in solution at electrode surfaces in iron cells, which are far greater than could exist in the bulk electrolyte perhaps in an interstitial state
- Adsorption of iron ions, and perhaps the salt compounds-themselves, within the carbon porous structure, which may be Langmuir or van der Waals processes, depending on whether they are attached as electrically charged components or as neutral molecules with dipole moments.
6.10 Further Examination of Concentration Ratios
In examining the cell from a practical viewpoint, it is obvious that most inorganic salt compounds are soluble in water only to the extent of 2 to 4 molar concentrations. If we evaluate the performance that can be expected from simple, two compartment concentration cells, depending on bulk concentration differences, the performance as practical storage cells is quite low.
For a concentration ratio of 10:1 of a divalent active species, such as sulfide, the cell potential is ~0.03 volts. A concentration ratio of 100:1 results in a potential of 0.06 volts. These are exceedingly low values for a device to be practical, and the major portion of the stored electrical charge would be delivered at substantially lower voltages than the above. The maximum charge transfer per liter of 3 molar solution to 0.03 molarity is only slightly more than the transfer from 3 molar to 0.30 molar.
In the experiments conducted that relate to this invention, potentials of 1.0 volt and higher are regularly attained and sustained. This development provides a mechanism for increasing the voltages and charge densities of a concentration cell by over an order of magnitude above that which are realized simply from differences in bulk electrolyte concentration differences of chemical species on opposite sides of a cell membrane separator.
The voltages obtained are associated with concentration ratios of well over 1000:1. An extremely important aspect of this development concerns the method of collecting and storing the differentials in concentrations at the electrodes. In the method employed here, the substances that produce the electric potentials are electrolytically produced ions injected into the micro-pores of both cathode and anode. Activated carbon, especially micro-pore coconut charcoal, is the most effective means for storing most molecular species in an available and reversible fashion.
The cell reagents are stored by means of adsorption of the van der Waals type. The carbon structure appears unaltered even after 10,000 cycles of charge/discharge events.
Activated carbon has a surface area of approximately 1,000 to 2,000 square meters per gram of accessible surface, depending on electrolyte conditions and physical properties of the solute to be adsorbed. Assuming about 103 m2, or 107 cm2 area per gm, and an average molecular area for sulfur or sodium polysulfide of 3 × 10–8 cm2, the number of molecules that could occupy the surface area of one gram of carbon as a mono-molecular layer is in the order of 1022 molecules, or about 2 × 10–2 moles. That number corresponds to about 0.1 ampere-hours of electric charge for a divalent species, such as sulfur.
The bulk density of the active carbon used is in the range of 0.60 gm/cm3, and its void space is about 60%. Hence, about 60% of the space occupied by 1 cm3 of charcoal would be filled by electrolyte in a concentration cell that employs such a material as the major electrode structure. From the preceding estimates, about 0.006 ampere-hours of charge can be stored per cm3 of electrode volume. The potential at the electrode surface is dictated by the concentration of the specific ion that is in the immediate vicinity (Helmholtz layer) rather than the bulk concentration that is some significant distance away. Returning to the above estimate of the number of moles adsorbed per gram of carbon, we find that the quantity of material in one cm3 of carbon is ~2 × 10–2 moles. That quantity of molecules, if dissolved in the 0.6 cm3 volume, would correspond to a 40 molar solution. In actuality, the electrodes encounter a much higher effective concentration because of the manner in which the species exists at the interface.
The necessity for introducing the specific reagents into the active carbon pores by direct ion migration by means of an electric field gradient in the charging process can be explained as follows. The rate of molecular diffusion, J, due to a concentration gradient is simply
(6.32)

where D is the diffusion coefficient, and c is the concentration of a particular molecular species in solution. Generally, D has a value of about 2 × 10–5 cm2 sec–1 for most salts in water solution. The thermal diffusion of solute into the pores of activated carbon would have to take place only as a result of the concentration gradient generated by the adsorption rate out of solution of the specific chemical species. That rate is quite slow. In effect, it would give rise to only a very small value of dc/dx in the above expression. The rate of adsorption of molecules from solution is directly proportional to the number of adsorption sites that remain available and the concentration of the chemical species in the immediate vicinity of the carbon surface. This is expressed by the familiar equation
(6.33)

where θ is the fraction of the total number of available sites that are occupied by the adsorbents on the carbon surface, and ka is an experimental constant of the tests. If the storage of a substance by adsorption depends solely on thermal diffusion as the driv ing force, the process is very slow and is severely counteracted by diffusion in the opposite direction as sites are increasingly occupied.
However, if an electric current is applied to the ionized salt solution with a current density of 0.01 amps/cm2, then the migration rate of the species will be in the range of 1016 ions per second, or about 3 × 10–7 gm sec–1 cm2. That is a much greater rate than can be expected from the thermal diffusion rate estimated above.
6.11 Empirical Results with Small Laboratory Cells
The voltages experimentally obtained in single cells do not conform to the simple expression (RT/nF) × ln(c1/c2). The values are much greater than predicted. The main question is why. Since the cell is symmetrical, voltages are kept low, and since there are no sources of energy other than concentration differences on opposite sides of the separator, the operation would appear to be a simple concentration cell.
Let us examine some of the cells’ performances. Doing so requires the following data:
- Cell electrolyte capacity ~ 12 cc per side
- Electrolyte composition and AH capacity to same composition on either side
- Some test data, i.e., AH input and voltages obtained
- Empirical potentials agree with the concentration relationship even when assuming no loss due to diffusion, etc.
Solutions have been 2.5 molar in Na2S with added sulfur of 100 gm per 100 cc of 2.5 molar monosulfide solution.
This would make the solution at full charge Na2S//Na2S4. However, this does not result in 0.05 AH capacity per 1 cc of total solution (both sides of cell). The empirical shapes of the charge/ discharge curves for the single cell-porous secondary cells do not conform to concentration cell mathematical predictions. A 10 cell module with no significant storage on porous electrodes did give the performance predicted by concentration cell math.
A significant question is whether the van der Waals “forces” provide additional energy for discharge in the porous electrodes, or whether it simply provides for a higher apparent concentration differential.
Also, there is the issue of the thermodynamics (energetics) associated with adsorption. Heat is usually liberated when molecules drop into the adsorbed state. If so, how does that affect the storage of energy? Usually, heat must be applied to free adsorbed state molecules. Hence, that cannot contribute to the cell potential. Perhaps it contributes indirectly by making the apparent concentration greater by holding onto species in greater surface densities. Is it possible that simply the huge increase in concentration as charging proceeds would account for this? Let’s assume that the electrolyte is the large “storage volume” of a cell for the reagents to reside in as a reservoir. Upon charging, these species are concentrated in very small volumes on the porous electrode surfaces and appear considerably more concentrated than the simple equations that employ the reservoir electrolyte volumes in the log relationship.
Generally, the energy (evolved) of adsorption is low, about 10 kcal/mole adsorbed, which is about the same given off when gas condenses to liquid. Thus, the energy of adsorption is to be subtracted from the potentials realized via concentration differences.
To explore single layer and multilayer adsorption, reviewing the Freundlich and Langmuir equations would be helpful. Langmuir relations apply best to saturated situations. Equilibrium is reached when desorption rates are equal to adsorption rates. One of the adsorbents can also be the solvent, taking up space in the adsorbent surfaces.
Now we will discuss what the electrode experiences in terms of electrochemical potentials within these layers. Is it strictly a concentration voltage, or are there other processes involved?
For treatments of energies of interfacial states, etc., see Moelwyn and Hughes, pages 922 et. seq.
Some factors that influence the shapes of curves of cell behavior are listed below:
- Electrical current, upon charging, “forces” the ions or molecules to become more concentrated than can be accomplished otherwise. The greater the current density, the greater the concentration of ion densities.
- As proximity of these ions increase, the greater are the van der Waal “forces” that tend to further concentrate them.
- As the population density of the species increases, the rate of adsorption increases at the electrode porous surfaces.
- At least the “apparent” concentration differences between cell electrodes are enhanced.
- We should address the reagent ion concentration seen by the electrode as the number of ions per unit volume in the immediate vicinity of the electrode rather than the calculations based on the “bulk” concentration of the cell on either side.
- The voltages observed across a cell are well beyond what the simple macroscopic calculations predict. They might then be due simply to a temporarily high build-up caused by slow reagent diffusion away from the electrodes during charging. Then, the dwell time of these voltages would be very much shorter than observed in laboratory cells.
If the above is true, then we must determine the specific volume for the electrode so that the concentration of reagents can be computed. So, let us see if we can approximate the performance of a cell as if it had small volume but molarity in the ranges of 10, 20, to 100. A good number to use for effective molecular diameters in these types of calculations, as obtained from diffusion and viscosity measurements, is about 5 × 10–8 cm. Then, examine the adsorption process on the basis of sorption rates due to thermal motion of molecules impinging on the outer porous electrode surfaces. Consider how movement under a voltage gradient impressed across the cell would change anything in terms of capture probability or desorption rates. For additional information about mean free paths and activation energies see pages 443, 448, and 348–350 of Daniels and Alberty. For some straightforward mathematical treatment of adsorption and capture probability see page 523 et. seq. Similarly, the Langmuir isotherms should lead the way for energy and electric potential relations in adsorption processes. Koreyta, in his book on electrochemistry, provides some inputs on pages 226, 227, (Helmholtz layer and Gibbs free energy of adsorption) and 246 on electrode processes and adsorption. He also discusses the solvent as being a structure less dielectric in which ions move about and interact.
Now consider the diffusion rate of a molecule or ionic species at room temperature within a solvent such as water. We can estimate this strictly from the classical kinetic theory. Then, see how many will diffuse across a unit area per unit time and compare this with the flow rate of an ion if a voltage gradient were imposed across that same area.
The diffusion coefficient D is defined in the relationship dq/dt = D (dc/dx), where the linear concentration gradient is in the direction normal to some unit area. The problem here in determining the amount of net flow, or number of molecules diffusing into the porous charcoal sites, is that there is no thickness layer to assign. Hence, we might just look at the desorption rate for the solid surface (and make a guess as to its surface volume) and solve for a diffusion layer thickness that would match adsorption rate data and diffusion. For example, if we plot the rate dc/dt for a given solution and molecular species and then look for where the curves of adsorption rates and diffusion rates intersect, it should tell us what the initial conditions are. As adsorption progresses, the rate of adsorption will decrease because sites are being filled and desorption becomes a significant factor in the ultimate equilibrium.
Moving boundary layer data with ionic solutions provides some useful insight into ion mobility as well as ionic velocities. The migration or diffusion of ions under the influence of a voltage gradient follows the same mathematical format as Fick’s law for molecular diffusion.
One simple configuration of a concentration cell employs the two soluble oxidation states of iron. In the case of metallic materials, we are much less interested in using their elemental state because of the consequent possibilities of short-circuiting a cell via metallic dendrites and gas evolution, especially of free metals on electrode surfaces in an acidic electrolyte.
The reaction of ferrous/ferric solutions discussed previously does operate very well as a concentration dependent cell, but care must be exercised during “charging” to maintain voltages below the hydrogen evolution level, usually in the vicinity of 1.2 volts in the acidic electrolyte. However, this problem can be avoided by resorting to non-aqueous solvents as electrolytes. Such cells employing organic solvents generally have higher electrical resistance, and the salts will usually be less soluble. Limited solubility is not necessarily a serious problem if the cells are designed to operate beyond that point with solids in the electrolyte.
6.12 Iron/Iron Concentration Cell Properties
Balancing the concentrations of components in a ferric/ferrous cell for the basic reactions that take place at either electrode is simply
(6.34)
The components on either side of a functioning concentration cell can be represented as shown below. The composition of the reagents must be the same on both sides of a cell separator at the end of discharge.
At the beginning of discharge (charged state), the conditions are 2FeCl2 // 2 FeCl3. The cell could be represented at the end of discharge as FeCl3 + FeCl2 // FeCl2 + FeCl3 symmetrical.
We can now calculate the maximum available energy density for this process, assuming a maximum of 1 volt potential difference in aqueous solutions to keep below the hydrogen evolution voltage in an acidic environment. The total molecular weight, ignoring that of water, is 2 × (56 + 2 × 35) + 2 × (56 + 3 × 35) = 574.
Since there is a single charge carrier, there is 26.6 AH per 454/574 = 0.79 lb of reagents. This works out to about 1 volt × 26.6 × 0.79 = 21 Wh/lb divided by 2 as an average load voltage, or about 11 Wh/lb or dry reagents. The actual number is quite a bit lower when accounting for weight of water, or other solvent, and the dead weights of electrodes and enclosure.
Despite the low ED, the system may be useful because of its low cost, high safety, and reliability of performance in stationary applications.
The above estimates presume that the electrical charge carrier is the chloride ion, Cl–.
A second version of this concentration cell could depend on positive ions as charge carriers. Perhaps the best would be the hydrogen ion, H+, because of its high mobility and consequent higher conductance in solution. A cation, or positive ion transfer membrane, could be employed in the cell for best separation and lowest diffusion losses. However, a micro porous membrane could serve well also.
The cell composition at full charge might look like 2FeCl2 + HCl // 2FeCl3. At the end of discharge, the components on either side of the separator would be the same with the exception that HCl has been transposed, or FeCl2 + FeCl3 // FeCl2 + FeCl3 + HCl.
The ED of this process is even less than that of the preceding because we must account for the additional weights of HCl in the reaction.
6.13 The Mechanisms of Energy Storage Cells
There are a few chemical elements and their compounds that lend themselves well to such processes. Preference at this time is for sulfur, and it is the material about which we have the most empirical data.
The reactions occurring at each electrode during both charge and discharge are ![]() .
.
The element and its alkali–metal compounds are cheap, plentiful, safe, well behaved at room temperature, and have very useful properties such as the ability to readily form “polymers.” These polymers are merely sulfur atoms attached to the sulfide components such as the following:
(6.35)

All of the above are very soluble in water and other polar solvents.
Please note, however, that the sodium ion has nothing to do with the potential producing mechanisms. It is merely the cation part of the compound that could just as easily be served by K, or NH4.
In order to have conduction within a cell and not have an accumulation of electrical charge on either electrode, the transport mechanisms must be accompanied by suitable oxidation/reduction processes for the exchange of electrons in an external circuit. Figure 6.13 below shows a very simple cell of two electrodes, an ion transfer separator, and two equal volumes, V, of electrolytes.

Figure 6.13 Cell diagram.
The cell voltage is proportional to the logarithm of the concentration ratios in the immediate vicinity of the electrode surfaces. However, for the sake of mathematical simplicity, let us assume that the cell potential, E, can be expressed as
(6.36)

Where
(6.37)

The simplified descriptive approach above avoids mathematical complexities that contribute virtually nothing to the main arguments of cell operation.
A plot of the voltage versus charge before substitution of the last expressions is a straight line, as depicted in Figure 6.14.

Figure 6.14 Simple linear dependence of voltage on concentration.
As the cell is charged from its totally discharged state, in which all concentrations are equal to Qo, more sulfides accumulate on one side (–), and the sulfide ions are depleted on the other (+) side.
However, if we substitute idt for the variable Q, the dependency of E upon Q is transformed from a linear dependency to a different shape. Making the substitutions, the subsequent approximations lead to the following equation for E:
(6.38)

To simplify further, we may just let i∆t = x to see how the parameters are interdependent. So, we obtain

where a also represents Qo.
Differentiating the above with respect to x,
(6.40)

A plot of E versus x looks like the illustration in Figure 6.15, in which the voltage changes increasingly rapidly as x increases and approaches the value a. This is hardly a linear relationship.

Figure 6.15 Shape of the functional relationship in equation 6.39.
It is obvious that, as x increases, the voltage is large for smaller intervals of x as x approaches a. But, at these higher potentials, the ion carrier population density grows quite small, meaning that less charge is available at higher voltages until x = a, when no further conduction is possible.
In practice, we would want to modify this behavior in a cell because only a small portion of the total charge would be delivered at the higher electric potentials. Herein lies the rather important, if not critical, part of this approach. First, we want to have as high a concentration of our reagents, in this case S–2 and S, as possible from the very start. This is partly accomplished by having some of the reagents Na2S and S initially in solid form within microporous carbon electrodes.
Remembering that only the sulfide ion S–2 counts in the math for determining potential, we want high concentrations of such ions along with the necessary sulfur molecules to be available at both electrodes. Normally, this availability is limited at any time because of the solubility of the salt, Na2S. Therefore, we store a fair amount of it in “solid form” at the electrodes.
Initially the cell has equal amounts of both sulfur and sulfides at each electrode. As charging progresses, sulfides are created at one electrode and sulfur at the other. A maximum value of sulfides at one electrode is reached, and continued charging results in solids coming out on that electrode stored for later discharge. Similarly, at the opposite electrode sulfur is deposited for later discharge.
When discharge begins, the cell voltage is quite high because of the concentration differential that develops. As the cell is discharged, some of the sulfides become solubilized, replacing the loss of sulfides on the concentrated side and, thus, keeping the potential up. On the opposite side during discharge, the transferred sodium ions that are produced result in continued diminishment of sulfide ions and the release of sulfur.
Thus, we have a reservoir of ions beyond solubility that is available to the electrolyte/electrode interface at some rate determined by diffusion constants and dynamics of electron transfer and solubilization rates. The discharge currents must be kept low enough so that the supply mechanism can keep up with the demands and maintain high potentials.
This is the case because inordinately high voltages have been obtained with cells that have been provided with such structures at the electrode surfaces. Cells with no porous carbon provisions generate potentials in the range of at most 0.03 to 0.08 volts even after prolonged charging. Smooth electrodes give the expected performance shown by the Nernst equation. The thermodynamics are certainly correct, but practical performance is immensely altered with storage, probably by virtue of interfacial forces of the van der Waals type.
The exact mechanisms of how the reagents are stored are not entirely understood, that is whether they are stored as solids adjacent to the electrode surfaces or actually adsorbed as ionic materials, etc. It makes a difference when one wishes to perform analyses or optimize parameters. Let us assume for the moment that, after solution saturation of the sulfides and polysulfides have been attained, further charging results in solids accumulation. Then we must consider the rates at which solution and dissolution takes place. Let us assume further that the rates with which the species are removed as solids and returned as solutes are simply proportional to the concentrations Ca and Cb of the respective species, sulfide and sulfur. Then the rates R at which the sulfides and sulfur solidify can be expressed as the following functions:
(6.41)

Obviously, the rate of removal of sulfide will be directly proportional to its concentration in solution, inversely proportional to the amount already in solid form at the electrodes, and to some extent due to the electrical current density. The exact form of the above functions can be better estimated with more modeling of the rate mechanisms.
The very reproducible and consistent experimental evidence that we have obtained to date with hundreds of cycles and many lab cells would indicate, according to the Nernst equation, that the electrodes are seeing concentration ratios as high as 1020 or more for us to be able to obtain 1.5 to 2.0 volts:
(6.42)

A fascinating aspect of this system is that it does not depend on the voltages of any couple such as Zn/Br (1.8 volts), Zn/Cu (0.8 volts), etc. The materials are the same on both sides of the cell only at different concentrations. Diffusion of unwanted materials from one side to another does not result in irreversible run down of the cell. Aging mechanisms are scant, no gasses evolve, and pH stays constant. The carnival prize seems to be worth the effort to squeeze maximum ED out of it.
The oxidation and reduction at the two electrodes necessary to get an electrical output is simply S + 2e = S=. Iron will do the same thing between ferrous and ferric.
The ionic species, sulfide ion, is stored within the porous electrode structure at effective concentrations that are much greater than what can be achieved in solution. The sulfide ion storage is at electrode surfaces. Some of that storage is probably in the form of solid sodium sulfide immediately adjacent to electrode surfaces and available to those surfaces as they return into solution. Some storage probably takes place in ionic form as adsorbed materials.
Charge retention is a function primarily of the solubilization rates of reactants and diffusion across a membrane barrier. Dependence on the latter should be minimized because of the unavoidably encountered trade off between molecular diffusion and electrical conduction.
6.14 Operational Models of Sulfide Based Cells
It is now necessary to develop some simple working models of the processes within a cell during both charging and discharging modes at each electrode. It will also be necessary to make some simplifying assumptions in our first attempt to explain what is happening at interfaces and at boundaries between phases.
There are only three forms in which the reagents can exist in the immediate vicinity of the electrodes: (1) as solids of composition Na2Sx and s, (2) in solution with water as Na+ and S= ions or dissolved as polysulfides (unionized sulfur attached to the Na2S molecule), and (3) in the adsorbed state on the porous carbon electrodes as ionic species or as polysulfides. This situation is illustrated in Figure 6.16 with some assigned designations for the concentrations of these various quantities. The diagram is strictly a schematic form and does not necessarily display the actual physical structure and relative locations of the components.

Figure 6.16 Sulfur species at electrode vicinities.
Certain additional presumptions must be made at this point in order to move forward with an analysis. These presumptions can and will be modified as we learn about what takes place inside the cell. When possible, we will take the path of greatest simplicity, Occam’s Razor approach.
As a first model for analytical purposes, let us assume only two nonporous electrodes and a membrane that separates the two compartments. This is shown in Figure 6.17 below.

Figure 6.17 Storage and flow model – first balance analysis.
The storage of reactants and reagents takes place in this first model solely in the electrolyte as dissolved components. The initial concentration, Co, of the active reagent, Na2S in this case, is simply
(6.43)

where V is the volume for electrolytes on each side of the cell. If we designate I as the electric current (ionic) passing through the cell upon charging, then the rate, dQ/dt, with which sulfide ions are produced from dissolved sulfur (polysulfides) in the (+) side of the cell is as follows:
(6.44)

There are four principal mechanisms that we know are involved in the operation of a cell. At the negative electrode during charging, they are the following:
- The generation of sulfide ions by migration of positive sodium ions toward the negative electrode
- The diffusion of ions across the barrier from one side of the cell to the other by virtue of concentration differentials of those specific species
- The adsorption and desorption of molecules onto and out of the surfaces of porous carbon electrodes
- As solute concentrations rise within an electrolyte, some portion will come out of solution (precipitate) as solids.
There are some other secondary issues associated with cell operation, such as concentration gradients within cell compartments, the number of water molecules adsorbed onto electrode surfaces with the solutes, and whether the adsorbents are still “in solution” as ionic species or as undissociated molecules. However, for the present we will ignore these matters to keep the operational explanations and descriptions as simple as possible.
6.15 Storage Solely in Bulk Electrolyte
To further shorten and make more specific the ensuing descriptions, let us treat the sulfur/sulfide system as the one in question, and let us be concerned, at the beginning, solely with the processes within the negative side of the cell. That then defines the species with which we are concerned – the S= ion and the S molecule. During charging, sulfur is reduced to sulfide with the acquisition of two electrons at the negative electrode. At the positive electrode, sulfides are oxidized to sulfur by giving up two electrons, a remarkably simple and symmetrical cell.
Looking strictly at the flow balance of S= ions generated at the electrode and diffusing across the barrier per unit area of working electrode and barrier, Ra = generation rate of S= ions = KaI, and Rb = loss rate by diffusion (Fick’s first law) = Kb(C1 – C2), where C1 and C2 are respectively the S= concentrations in the negative and the positive sides.
Net rate, R, of accumulation of S= in the negative side is
(6.45)

Putting the above in terms of electric current and ion quantities, we get the following: at any time, t, the amount of S= ions present in the negative and positive sides are Q1 and Q2, respectively, and the sum of these amounts is always Qo. Thus,
(6.46)

Initially in a discharged cell Q1 = Q2, and as the cell is charged by the electric current Q1 becomes larger until it reaches a value great enough such that the rate of charging equals the loss of S= ions via diffusion across the barrier. This value is attained when Ra = Rb, or
(6.47)

The concentration differential attainable in such a simple cell is severely limited by diffusion losses, and the cell potential is limited by the maximum concentrations that can be provided for the electrodes. The total electric charge available at higher cell potentials would be quite small, as is evidenced by the algebraic comparisons below of charge density at different voltages.
Since the sum of the sulfide ions present within the cell is a constant, we shall designate it Qo. Then, it follows that Q1 + Q2 = Qo. As Q1 approaches Qo, Q2 approaches zero.
If we continue, for the purposes of mathematical simplicity, in letting the cell potential, Ec, be directly proportional to the cell concentration ratios rather than its more representative form given by Nernst, (the relationships will be corrected later after the present arguments for dynamic cell balance are established) then we can state the following:
(6.48)

Upon charging or discharging the energy, ψ, we can evaluate the integral between two different sets of limits and compare the numerical results for energy input or output over the respective voltage ranges.
The energy over any interval of time or voltage may simply be represented as
(6.49)

Expressed in a more useful manner, the incremental change in energy because Ec dQ = Q dEc is
(6.50)

The integral of this equation is
(6.51)

Putting the above expression in terms of voltage, we obtain the more convenient form
(6.52)

And the differential of energy assumes the form
(6.53)

Integrating this last expression in terms of E, we get
(6.54)

Now, to illustrate that there is less energy available over any voltage interval upon discharge at the higher voltages, we need to merely look at the derivative of ψ to see that as E becomes very large the slope of the curve of ψ decreases and approaches a constant value of Qo.
At a later time, the more representative Nernst expression can be substituted for cell potential, E, i.e.,
(6.55)

At present, the exact form of the voltage dependence on concentrations is not important to establishing the argument for dynamic stability of the cell.
6.16 More on Storage of Reagents in Adsorbed State
In our previous approach to describing a dynamic balance of ion flow we considered only the rate of species generation via electric current counterbalanced by the loss of those species across to the opposite side of a cell by diffusion through a membrane. Membrane impedance to diffusion loss is limited mainly by a compromise with electrical conductivity through the same separator.
Let’s introduce an additional and primary method of materials storage – adsorption. This is accomplished by employing microporous carbon attached to the surface of both electrodes. In the negative electrode, S= ions are stored in the form of the Na2S compound, and in the positive electrode the S is stored as either Na2S unionized, Na2Sx, or as the element S. Actually, S is stored at both electrodes during the entire process as a reservoir of reagents during charging at the negative electrode and for discharging at the positive electrode. However, the presence of S at either electrode does not enter into the thermodynamic relationship for electric potential.
With such a modification of cell design we are able to develop much higher effective ionic concentrations. Let me use a fictitious configuration below to illustrate the essence of such improvement.
If it were possible to establish a very small compartment with volume, δ, per unit area immediately adjacent to each electrode by means of an idealized barrier, as shown in Figure 6.18, we would be able to build electrolytically gigantic concentrations of species supplied by the much larger reservoirs of volume V.

Figure 6.18 Hypothetical compartments at electrode surfaces.
From our earlier relationship regarding potentials and concentration ratios (we will stay for a short while longer with the linear model rather than the more representative logarithmic one), the cell potential as a function of charge now becomes associated only with the concentrations in the region δ. Before we can proceed further with modeling the cell it is necessary to more closely examine the processes at the electrodes within the δ region. (See Figure 6.19).

Figure 6.19 Representation of processes at electrode surface regions.
In the previous simple situation of flat electrodes and a single membrane, we assumed perfect and instantaneous mixing of electrolyte solutes. If we make that assumption again in this new model, we encounter some difficulties. Let us look at what happens at the negative electrode (realizing that a similar situation takes place at the positive side). In order for the reduction of sulfur to sulfide ions to occur, it is necessary to have a continuous supply of sulfur immediately available at the negative electrode. This supply can exist as solid sulfur in the electrode or as polysulfide ions in solution. In order to be able to charge the (–) electrode, sulfur must have either been previously stored within the δ compartment, or it must be made available as a stream from the large V reservoir.
A mechanism is needed to keep the S= ions in the immediate neighborhood of the (–) electrode in order to retard their drifting away into the bulk solution.
The latter explanation is based on the small compartment having free access via molecular diffusion to the entire electrolyte. That would make the situation identical to “storage in bulk electrolyte,” as described in the first case. In addition, high concentrations cannot be accumulated because of mixing. Then, other than the storage of sulfur prior to charging, there is no method we can visualize in which the δ region can sustain the high concentrations necessary for high electrode potentials.
However, it is possible to accumulate extremely high sulfide concentrations by adsorption within the electrodes during the charging process. An example of high surface area carbon shows that as much as 2,000 square meters of area is available per gram of activated carbon. Looking at what that might mean in terms of molecules of storage capability, we perform the following quick approximations. Since the average, effective molecular diameters are in the order of 2 – 5 × 10–8 cm, the average area of small molecules is in the order of 10–15 cm2. The available area of activated carbon surfaces is 2 × 103 m2 = 2 × 107 cm2. Dividing the two numbers gives us about 2 × 1022 molecules or atoms stored per gram of active (microporous) carbon.
An interesting note here concerns the wall thickness of the porous carbon. One gram of carbon is 1/12 of a molecular weight. Since there are about 6 × 1023 molecules per gram equivalent weight of substance (Avogadro’s number), about 5 × 1022 molecules of carbon are present to do the adsorbing, or about 3 molecules of carbon per molecule of adsorbent. If we further assume an agreement with the Langmuir model of molecular surface bonding via a Van der Waals type of force, then the adsorbent is a monomolecular layer, and the carbon wall structure is on average not much more than 2 to 3 carbon atoms thick.
The average specific gravity of such activated carbon is in the range of 1 gram per cm2. Then we see that about 2 × 1022 molecules, or ionic species, are stored per cm3 volume of porous electrode. To find the storage capabilities of this electrode in terms of electrical charge (amp-hours), we look at the relationship provided by Faraday. The Faraday equivalent is 96,500 coulombs per equivalent, or about 105 coulombs per single valence molecule. Since sulfur has two charges, that number becomes 2 × 105 coulombs.
Since there are about 2 × 1022/6 × 1023 = 0.03 Avogadro numbers of S atoms stored, it requires in the order of 0.03 × 2 × 105 = 6 × 103 coulombs to electrolytically transport that number of dual charged ionic species across the cell. That number is 6 × 103 ampere seconds, or about 1.7 ampere-hours of charge (per cm3 of electrode).
That is a fairly high charge density for an adsorbent electrode. There is significant evidence that adsorption of molecular species in solution will occur in more than one molecular layer (Freundlich and Langmuir’s adsorption isotherms).
Now we can return to the consideration of rate processes. Superposed on the previous situation of bulk storage, the rates of adsorption/desorption must be taken into account. Again, as a first approximation, let us use the Langmuir expression regarding adsorption isotherms. As discussed earlier, this approximation does not account for changes in adsorptivity as the surface sites become more occupied, and the ratio of the coefficients αa and αd, the adsoption and desorption, in the relationships of previous equations is not constant.
(6.56)

If we re-examine equations (6.25) and (6.26), perhaps we can put all the variable quantities of S= ions in terms of the amount adsorbed, Qa. We know that at time zero the following conditions exist:
- Qa = 0
- P = 0
- C1 = Co
- Cδ = C1 = Co
- Q1 = Qo
Also, at t = 0, the rate of change, or increase of Qa is
(6.57)

We can next proceed to conclude that the electric potential of the cell can be approximated as a function of the ratio of adsorbed species on each of the two electrodes, or
(6.58)

6.17 Energy Density
An interesting estimate is the energy density available from a cubic centimeter of elemental sulfur in the charge transfer of one volt.
The specific gravity of the sulfur atoms is about 2 grams per cm3, and its atomic weight is 32. With two electrons per atom transfer upon oxidation/reduction, about 100,000 coulombs × 1/32 × 2 would be the charge exchange per gram of sulfur to S=. However, since 2 grams are present per cm3, the math works out to about 1.2 × 104 coulombs per cm3.
Since 1 coulomb is an ampere-second, the charge transfer is about 3 amp-hours, which corresponds with 3 watt-hours of energy. Since the cells are symmetrical and there is an equal amount of material on either side, these numbers need to be divided by 2.
Accounting for both sides of the cells, this works out to a maximum energy density, assuming an average potential of only 1 volt of about 24 Wh per in3, or over 41 kWh per cubic foot of cells. That is a very respectable number, even neglecting weights of water, electrodes, case, etc. Also, these cells might be able to operate at levels of many volts, thus increasing the energy proportionately.
6.18 Observations Regarding Electrical Behavior
To provide a general idea of the shape of some typical charge/discharge curves, the following graphs (Figures 6.20–6.25) qualitatively show how these cells behave. Actual data with current and voltage values are supplied later in the book. For the present, these do show the nature of the cells and clearly show that they do not behave in any linear fashion. The discharge curve for constant current, constant load, or any other control is not “flat” with time. Efficiency of charge and discharge can be very high if the difference between open circuit and charging/discharging potentials are maintained small and, perhaps, constant as diagrammed in the last drawing.

Figure 6.20 Voltage limited charging.

Figure 6.21 Constant current charging.

Figure 6.22 Fixed load discharge from “overcharge”.

Figure 6.23 Constant current discharge.

Figure 6.24 Maintaining a constant charge to open circuit differential.

Figure 6.25 Discharge at constant power output.
The power supply and load should be designed to follow these curves for best efficiency.
6.19 Concluding Comments
The following is only a general outline of the basic rationale behind the development of concentration cells. These types of cells do offer a new and different class of phenomenon that can be engineered into practical devices for the storage of energy. Even though the above discussion is centered on the element sulfur, there are many other compounds that could serve the same purpose. Sulfur has ebb, chosen here primarily because it has been experimentally studied most extensively and because its physical and chemical properties lend themselves to experiment easily. There are no problems with high oxidation rates, toxicity, solubility, etc., with which to cope. Iron will serve the same purpose as sulfur. The ferric, Fe3, and the ferrous, Fe+2, states are analogous to the S= and S states of the elements.
Iron poses one problem of inconvenience in the laboratory, which is its tendency to oxidize in the presence of air. However, that is certainly not a problem in the development of practical hardware.
In order to achieve high working potentials of over 2 volts, a non-aqueous electrolyte must be employed to avoid the decomposition of water during cell charging. Again, that is not a problem in a reasonably well-equipped laboratory. There are numerous stable and compatible non-aqueous solvents that can be employed with good conductivities. One concern is the necessity for the occlusion of air and, particularly, water vapor because most of such solvents tend to be quite hygroscopic.
The diagram shows both cations, M+, as well as anions, X–, migrating from the respective electrodes with opposite electrical polarity as charge carriers during the charge mode. Upon arrival at the (–) electrode, the Ma ions are reduced to Mb form and acquire electron(s) from the power supply in the external circuit. In the positive electrode side of the cell X–, ions associate with the Ma ions, whose charge is greater formed at that surface.
During the discharge mode, exactly the opposite of these processes occurs, and stored electrical energy as complex concentration differences in X– and M+ are restored.
6.20 Typical Performance Characteristics
The plot in Figure 6.26 shows typical test bench results of small engineering cells with an electrode area of 10 square inches. The curve is for the discharge mode of operation at constant power delivery to a load. Total charge capacity of the cell in this instance is about 0.40 amp-hour.

Figure 6.26 Volt-amp data.
Voltage and current are continuously changing to maintain constant power at 0.10 watt. External power management circuits are employed to achieve this type of performance.
6.21 Sulfide/Sulfur Half Cell Balance
The information contained in the following text, graphs, and mathematical development concerns the properties of a symmetrical electrochemical cell employing the basic and reversible reaction at both electrodes. The electrolyte is an aqueous or other suitable solvent such as alcohol or a solution of an alkali sulfide salt such as (NH4)2S, Na2S, or K2S. Since such sulfide salts solubilize sulfur, there are no solids present during normal operation of the cell.
A microporous membrane with ionic selectivity is employed as the separator between (–) and (+) electrolytes. In this instance, both Na+ as well as S–2 ions migrate in opposite directions as dictated by the electrical polarity of the electrodes. The rate of such transport for these ions is determined by their respective mobility through the solutions.
The polysulfide Na2Sx is the state common to both sides of the cell at the totally discharged stage. There are m-moles of each compound in solution on each side. When charging begins, higher polysulfides are generated at the (+) electrode, and lower sulfides are produced at the (–) electrode. If posilytes and negalytes have equal volumes, then at full charge the (–) side will have a saturated solution of the maximum solubilized sulfur, or Na2S5, as electrolyte, and the (+) side will be a maximum concentration of Na2S electrolyte.
If the concentration and/or volume of the (+) side is greater than that of the (–) side, then some free, solid sulfur may be deposited onto the (–) electrode surfaces at full charge. Such a configuration results in an increase in energy storage capacity of the cell since there would be less sodium and, perhaps, water needed for cell operation.
6.22 General Cell Attributes
The primary reason for pursuing a cell of this type is its potentially long life and maintenance-free operation. Since both sides of the cell contain the same chemical species, there is no possibility of degradation of performance or structure with time or cycling. The electrical potential in the cell is derived from the difference in concentration of one chemical species. In this instance, it is the sulfur/sulfide couple. Initially (cell in the “discharged state”), the concentrations of sulfur and sulfide ions are the same on either side of the cell. Each side is separated from the other by a microporous or ion selective membrane.
Attributes of the cell include the following:
- Benign chemical environment
- No maintenance
- Unlimited shelf life
- Unlimited cycle life
- No gas production
- Versatile system for either sealed unit or circulating electrolyte designs
- Very inexpensive and abundantly available chemical reagents
- Simple and inexpensive cell construction
It would seem that the advantages of indefinitely long life, abuse withstanding, and low cost would more than compensate for the above limitations. The high dependence of cell voltage on the state of charge is not different from that of employing compressed gas, rotating mass (flywheel), or capacitive storage since their working potential is similarly dependent on the amount of stored energy at any moment in time.
6.23 Electrolyte Information
The three salts cited earlier have high solubility in water and in alcohol. Some data is provided in Table 6.3.
Table 6.3 Salt data.
| Salt | Molecular wt. | Solubility | Resistivity |
| Na2S | 78 | 200 to 500 g/L | 4 to 8 ohm-cm |
| K2S | 110 | >800 g/L | 3 to 6 |
| (NH4)2S | 68 | >1000 g/L | 6 to 10 |
From: Handbook of chemistry & physics, 1981, CRC Co.
Voltage produced within the cell is due to the concentration ratio of chemical species at the electrodes and does not depend on the absolute concentrations of solutes. It is important to operate cells at high salt concentrations in order to minimize water, electrode, and cell structure weights, and to maximize energy density.
Consider a cell that employs a near optimized electrolyte composition such that all materials balance in the ion transfer processes. If we wish to keep all components in solution, then the electrolytes are as follows at the beginning of discharge: (–) side Na2S5 // 5 Na2S (+) side.
As discharge proceeds (assuming only Na+ ions are transported), compositions of each cell side come, as indicated in the following steps, all the way up to reverse total charge. Table 6.4 shows how the cell potential decreases as discharge progresses and how the electrical polarity reverses if carried further by an external power supply. The flow of electrons during “discharge,” in such a symmetrical cell, is from the negative electrode to the external load. When zero potential is reached at the same concentrations of S and S=, the flow of electrons goes to the negative electrode from the power supply during the “reverse charge” shown below.
Table 6.4 Cell potential decreases.
| Cell potential | (–) Side | (+) Side |
| 0.02 volts | Na2S5 | 5Na2S |
| 0.09 | Na2S4 + Na2S | 3Na2S + Na2S2 |
| Discharged | ||
| 0.00 | Na2S3 + 2Na2S | Na2S3 + 2Na2S |
| Charging | ||
| –0.09 | Na2S2 + 3Na2S | Na2S4 + Na2S |
| –0.02 | 5Na2S | Na2S5 |
There are 26 AH of charge per liter per gram equivalent weight. Thus, there are 156 AH transferred for the 6 moles of sodium ions. This corresponds to 0.156 AH/cc of total electrolyte, or about 9.36 AM per cc. For a cell with 1 in2 electrode area and a total spacing of 0.020 in, its electrolyte volume would be 0.020 in3 = 0.328 cc. This cell would then have a charge capacity of 9.36 AM/cc × 0.328 cc = 3.07 AM.
Higher capacities can be achieved if the polymerization of sulfur can proceed further or if free sulfur is allowed to accumulate.
Also, there are additional small voltage contributions from the formation energy of the polysulfides. For example, the following are some measured values from Oxidation Potentials by W. M. Latimer:
(6.59)

and
(6.60)

which gives differences of 0.01 volts per “polymerization” stage.
If we were to postulate that sulfur would polymerize to the S7 state, we would have a series of concentration and corresponding oxidation exchanges such as the following:
(6.61)

However, it is not necessary to rely on polymerization greater than 4 or 5 for sulfur since any sulfur on the (–) side would eventually be solubilized as the cell discharges. The first chemical equation of the above group could be written as
(6.62)

with the same performance results. As a matter of convenience, and to reduce the number of characters in these chemical balance equations, the sulfur is all expressed as polymer attachments.
The manner of implementing this increase in AH capacity is via plating out free sulfur if necessary and increasing concentration or quantity (volume) of the polysulfide side at the beginning of discharge. As one can see, if there is an excess (stoichiometrically) of polysulfides on the (–) side, then free sulfur can be generated on the (+) side during charge, thus giving rise to a very small concentration of Na+ and S–2 ions in that electrolyte. The resultant concentration ratio of ions across the separator would be very large. However, such a large ratio would be dissipated quickly upon discharge, as will be seen in the ensuing equations.
6.24 Concentration Cell Mechanism and Associated Mathematics
For any chemical reaction of the common form
(6.63)
the lower case letters are the numerical ratios of the upper case reactants.
The free energy change for the reaction is expressed as the following:
(6.64)

Or, it can be represented as
(6.65)
in which K represents the equilibrium constant for activities in the equilibrium state, and Q represents the activity quotient or ratio of activities of the products to those of the reactants. Since, and if reactants and products are at unit activity, ln(Q) = 0 and E = E0. Thus, E = E0 – RT/nFln(Q).
For most situations of interest here, the expression becomes
(6.66)

where aA and aB are the activity coefficients for ions A and B. For those conditions encountered in these electrochemical cells, the ratio of activity coefficients is equal to their concentration ratio, or in this instance of a sulfide/polysulfide concentration cell,
(6.67)

In addition, we have the potential due to the sodium ion concentration differences, or
(6.68)

where C1 and C2 are the concentrations of the same ionic species on each side, respectively, of a cell.
Now, let us examine a specific cell configuration for analysis and study. We will choose a unit cell with an active frontal area of 1 in2 and an electrolyte thickness of 0.010 inch on each side of the separator. This cell has a total electrolyte volume of 0.020 in3 × 16.4 cc/in3 = 0.328 cc.
An initial solution of concentration of 9 molar has Na2S in the (–) side and the equivalent of Na2S9 polysulfide in the (+) side. These may be in the form of undissolved monosulfide and undissolved sulfur on the (+) electrode.
6.25 Calculated Performance Data
Returning to the expression for concentration potential, we must relate the concentrations to the specifics of the cell volume and the dynamics of electric current flow. In general, the concentrations C1 and C2 may be written as C1 = Q1/V and C2 = Q2/V, where V is the volume on each side of the membrane.
During discharge at any time, t, after full charge, the quantities of reagents in each side are as follows:
(6.69)

The reason for normalizing the performance model is to relate the discharge rate to capacity, cell resistance, voltage polarization rates, and eventually diffusion limiting considerations.
Thus, our model is a one square inch area configuration with electrolyte spacing of 0.010 inches. Its volume is 0.328 cc. Concentration of initially charged solution in the (–) side is 9 molar Na2S. The transfer of 18 normals of Na+ ions would give a total of 28.1 ampminutes per cc of total solution (Table 6.5). The (+) side has a concentration of 1 molar 2Na+ ions corresponding to 28.1/9 = 3.12 AM/cc. If this is indeed the initial condition of the cell at the start of discharge, then we can set up a mathematical relationship to evaluate voltages as discharge progresses.
Table 6.5 Charge equivalents.
| Charged state | AH/L | AM/cc | AM/0.328 cc |
| (A) Na2S5//5Na2S | 260 | 15.6 | 5.12 |
| (B) NaS//7NaS | 364 | 21.8 | 7.15 |
| (C) Na2S9//9Na2S | 468 | 28.1 | 9.2 |
| (D) Na2S11//11Na2S | 572 | 34.3 | 11.3 |
Now, it is necessary to set the initial concentrations and then calculate potentials versus discharge time.
Then the ampere-minute quantities in the 0.328 cc volume cell become
(6.70)

The complete expression for the cell potential at various stages of discharge is determined by
(6.71)

where we have employed 0.02 amp as constant discharge current, n = 1 for sodium ions, and operation is at standard temperatures.
This expression has been programmed into a Lotus spreadsheet and a sequential method was employed to estimate voltages, energies, and power output during discharge. Since there is some uncertainty as to the sources of voltages between concentration ratios of Na+, S–2, and the formation of polysulfides during concentration changes, we will look at the type of performance obtainable for different assumptions.
Empirical data is available for single cell in shallow cycling operation. The rather high potentials realized at early stages of charging suggest that there are numerous mechanisms present as well as some significantly steep concentration gradients established at the surfaces of electrodes, even at low current densities.
In attempting to estimate performance via mathematical modeling, we have choices to make regarding the range of molarity change of ionic species, the initial concentration ratio (depth of dilution of sodium ions), and whether a multiplier is appropriate to the voltage equation in order to account for the observed shape and magnitude of empirical voltage discharge curves.
Three different cases (A, B, and C) will be selected for graphing. Case (A) is a situation of least energy capacity, where the multiplier = 1, 5 molarity of Na2S, and initial ratio of concentration is 5:1.
The equation describing the voltage versus discharge time is
(6.72)

depending on the extent to which the cell is charged beyond its above stipulated stoichiometric ratios.
Case (B) involves a mid-range energy density cell, where multiplier = 5, and m = 7 molarity of Na2S, and initial concentration ratio = 70:1.
Based on experimental evidence, it seems that a multiplier of value 5 for the entire concentration potential equation is reasonable and consistent with experimental results. In addition to the energy of formation of the polysulfides, concentration differentials are generated within the cell for sulfur, sulfide, and sodium ions.
The figures to follow are graphical representations of a cross section of numerical cell value combinations of the factors discussed above.
The more general equation for voltage may be given as
(6.73)

We arrive at the initial concentrations of the (–) and (+) reagents as follows. If the ratio desired is R, and the increment, d, is added to the (+) side, at the decrement of the (–) side is
(6.74)

Hence, if we wish a ratio of 70:1, about 10 times as that obtained, and if the concentrations were 5 molar and 1 molar, respectively, then solving the above for d gives 70(1.02 + d) = 1.02(7) – d, and d=0.905, and the new equation for voltage is
(6.75)

Case (C) involves a high capacity cell configuration, with multiplier = 5, an 11 molarity (+) solution, and an initial concentration ratio = 1100:1.
The values calculate as follows: 1100(1.02 + d) = 1.02(11) – d, and d = 0.99, and the voltage, E, becomes
(6.76)

6.26 Another S/S–2 Cell Balance Analysis Method
Perhaps another more direct and simple method of showing the materials balance and estimating the energy density of a concentration cell is that shown below for the sulfur/sulfide cell. We can assume that the process will no longer be limited to the maximum amount of sulfur that the polysulfide can solubilize. As an idealized example, the initial condition for a fully charged cell is Na2S // S, or more generally aNa2S // bS, where a and b are whole numbers of moles. In order for the process to balance at zero charge (complete discharged) state, a = b, and a > 1.
We can now compute the maximum charge stored per unit weight of reactants in this concentration cell. The simplest example would be 2Na2S // 2S at full charge, and Na2S + S // Na2S + S at total discharge, with a transfer of 50 AH per total molecular weight of reactants.
This amounts to 2(78) + 2(32) = 220 gm with a charge transfer of 50 AH giving as energy density 50 AH/220 gm × 454 gm/lb = 103 AH/lb of dry materials.
It is possible to further generalize the analysis for the cell processes wherein the sulfur is always attached to the sodium polysulfide molecules. Since the details of interim stages of complexing can be readily known, we will assume the following steps in the charge transfer and discharge of a cell that begin with the polysulfide on one side and the monosulfide on the opposite side. Let us take the pentasulfide as the largest size complex available. The cell configuration and reactions become those shown below.
Starting with the fully charged state as before, but with the bisulfide on one side and the monosulfide on the other, aNa2S // bNa2S2.
The smallest value for a is 3 since it is necessary to remove two 2Na atoms from the monosulfide in order to meet the conditions of no free sulfur on either side of the cell. Without going through the approximation sequences, the numerical ratio that results functions to make both sides of the cell identical after discharge is a = 3, b = 2, 3Na2S // 2Na2S2 Charged, and Na2S + Na2S2 // 2Na2S + Na2S2 Discharged.
The total gram molecular weight of both sides is 234 + 220 = 454, and the charge transferred by 2Na+ ions is 50 AH. The charge density of dry salt is simply 50 AH × 454 gm/lb /454 gm = 50 AH/lb.
If we start out with the trisulfide, the reaction balance, etc. are aNa2S // bNa2S3 Charged, a = 3, b = 1, 3Na2S // Na2S3, and Na2S + Na2S2 // Na2S + Na2S2.
Total weight = 3 × 78 + 142 = 376, and the charge density = 50 AH × 454/376 = 60 AH/lb.
The cell reaction making use of the next higher initial polymer of sulfur and sulfide is as follows: aNa2S // bNa2S4, a = 5, b = 1, 5Na2S // Na2S4, 3Na2S + Na2S2 // Na2S + Na2S3, and Na2S + 2Na2S2 // 2Na2S + Na2S2.
Total weight is 390 + 174 = 564. Since there are four Na+ ions transferred from fully charged to symmetrical distribution of ions at discharge, the charge density is 100 × 454/564 = 80 AH/lb.
Taking the pentasulfide as the last or highest complex, the cell parameters become aNa2S // bNa2S5, a = 7, b = 1, 7Na2S // Na2S5, 5Na2S + Na2S2 // Na2S + Na2S4, 3Na2S + 2Na2S2 // 2Na2S + Na2S3, Na2S + 3Na2S2 // 3Na2S + Na2S2.
Total weight is now 546 + 206 = 752. There are three transfers of 2Na+ ions, hence the charge density is now 150 AH × 454/752 = 90 AH/lb.
If it were possible in a practical cell to utilize higher complexes, the charge density would merely approach the maximum value of 103 AH/lb. In order to compute the energy density of such cells, it is necessary to multiply the charge density by an appropriate voltage. Since the cell potential is so dependent on the state of charge, a reasonable value of working cell voltage over the entire range of charge storage would be half of the full open circuit voltage of 1.0 to 1.2 volts, or about 0.5 to 0.6 volts. Hence, the maximum energy density of the cell, assuming no water (solvent) weight or other contributions to inefficiencies, would be about 52 to 62 WH/lb of reactants.
The operating open circuit potential is purposely limited to between 1.0 and 1.2 volts to prevent the evolution of hydrogen gas at electrode surfaces. H2 evolution would necessitate the periodic readjustment of electrolyte composition and the venting of cells and would eventually result in mechanical erosion of electrodes. Another approach to preventing gas generation at electrodes is the employment of non-aqueous solvents such as absolute alcohol, pyridine, DMSO, and nitriles.
6.27 A Different Example of a Concentration Cell, Fe+2/Fe+3
The principles employed in concentration cell design are not restricted to the use of sulfur and numerous polysulfides such as those of potassium, ammonium, lithium, etc. In fact, the above concentration cell approach to energy storage can make use of numerous other materials that have properties suitable to practical methods of implementation and different characteristics that may make them more applicable to certain uses. These materials include the use of the elements iron, bromine, iodine, and chromium. Their behavior as electrochemical species is well known and readily available.
The balance relations for the iron concentration cell are as follows. We will make use of the two oxidation states of iron, Fe2 and Fe+3 ions. Their solubility is such that high concentrations (two to four molar, respectively) of these are easily attained in water. Potentials during charge must be kept below that for the formation of free iron, Fe0.
That potential in water solutions is about 1.2 volts. The reaction of interest to us here is of the form aFeCl2 + bHCl // cFeCl3 + dHCl fully charged state.
The charge carrier is the hydrogen, H+, in this cell. A cation exchange membrane, or a microporous separator, is employed in this cell.
In order for the reaction to proceed and have a symmetrical situation on both sides of the separator, i.e., no further oxidation/reduction energetics remain, the minimum values for the coefficients a, b, c, and d are 2, 1, 2, and 1. Thus, the initial and final states are 2FeCl2 + HCl // 2FeCl3, and FeCl2 + FeCl3 // FeCl2 + FeCl3 + HCl.
There is only one charge carrier (exchange) per such step. Hence, the total weight of reagents is 252 + 36 + 322 = 910 gm. The charge density is then 25 AH × 454/910 = 13 AH/lb.
Even though the energy density is not as attractive as that of the sulfide system, there are some outstanding features such as extremely low cost of materials, low hazard, and no chance of solids deposition if potentials are kept below that for Fe+2 + 2e– → Fe0.
6.28 Performance Calculations Based on Nernst Potentials
A series of graphs and calculated data plots has been generated based on a simplified model of the volt-amp behavior of a basic cell. This was done in order to acquire a preliminary appreciation of the type of behavior that can be expected from actual cells, which were simultaneously fabricated and laboratory tested. Such calculations, along with graphs of the voltage versus current-time or state of charge of a cell, enable us to determine whether our actual test cells are operating in an expected fashion, or whether there are some discrepancies between actual behavior and our simplified understanding of the mechanisms involved. Even if the data might be quantitatively different from that calculated, the main importance in these types of investigations is whether the qualitative aspects are as predicted, or in other words, are the shapes and general behavior of the two groups of data, i.e., the calculated versus the empirical curves.
Despite the absence of many factors in the modeling such as diffusion and coulombic inefficiencies, it was found that the agreement between the two is quite close. To best illustrate these initial attempts to analytically represent cell performance, a few simplified models follow.
6.28.1 Constant Current Discharge
We will now look at the behavior of a cell when confined to discharging at a constant current by providing a suitably varying electrical load at its terminals. The mathematical formula for the value of cell voltage as it varies with the state of charge, on which the plot is based, is given as
(6.77)

where the terms a and b are the concentrations of the active reagent in the opposite sides of the cell. In this case, that reagent is the sulfur ion, S=. The subscripts n and n–1 are employed to designate the sequence in time of the terms. For example, the voltage at any time, t, indicated by subscript n is found from the value of an, which occurs at that same time, and the value of an–1, which occurred at the previous time interval. This arithmetical approximation enables us to make the evaluations in a spreadsheet program such as Lotus or Excel. The evaluations can thus be made piecewise, and plots can be generated from the calculated data. It is also assumed here that the volumes on either side of the cell are the same and normalized to give a one-to-one correlation between AH and change in concentrations.
To complete the mathematical description, the relationships for the a and b terms are as follows:
(6.78)

A constant current of 0.05 amps is almost arbitrarily assumed here per square inch of electrode surface area, and the time interval in obtaining readings of voltage is expressed as tn – tn–1 in the expressions (6.78).
The amount of electrical charge, Qn, removed from a cell over a time interval tn – tn–1 is given as
(6.79)

Figure 6.27 is such a graph where the cell potential, E, is plotted against the percentage of remaining electrical charge within the cell. A distinct disadvantage of concentration cells is the high dependence of voltage on SOC. However, this characteristic can be largely overcome, as discussed earlier in this chapter and later in Chapter 8, by having most of the reagents (sulfide ions) in the solid state at the electrodes with controlled dissolution as charging or discharging progresses, depending on which electrode polarity is involved.
Figure 6.27 Calculated – voltage versus remaining charge.
6.28.2 Constant Power Discharge
Another informative graph that shows the behavior of cells, when operated to deliver constant power during discharge, is obtained from the simple assumptions and relationships shown below. In these analyses, the predominant shape of the curves is logarithmic because of the fundamental equation for voltage as set forth in Nernst’s equation. As before, the voltage is proportional to the log of the ratio of concentrations of specific ions on each cell side, or
(6.80)

Similar to equations (6.78) and (6.79), the expressions for a, b, and Q now are as follows:
(6.81)

The total electrical charge that has passed through the load at constant power of course is
(6.82)

where N is the total number of samples taken over the time interval of interest. A chart of such a discharge is shown in Figure 6.28.

Figure 6.28 Voltage versus discharge time @ constant power.
The major portion of the electrical energy, or charge, is delivered at the lower levels of voltage because of the very rapid decline of the ratios a/b from fully charged. The only method we know of to reduce this rapid decline is the introduction of a stabilizing feature. This would maintain high a/b values by storing the major portion of charge in the form of unionized (solid) reagents at the electrode sites and then gradually replenish the ionic reagents by controlling their precipitation and dissolution at the respective electrodes.
6.29 Empirical Data
Countless laboratory cells and arrays based on concentration differentials have been constructed and tested. Most of these test cells were either sulfide or ferric/ferrous based chemistry systems. Numerous other systems such as bromine/bromide, iodine/iodide, and cupric/cuprous cells have been explored as well. The electrical results, as one would expect, obtained are very similar to those with the sulfide devices. However, other problems were encountered with these and other alternative cells associated with chemical stability (attack of surrounding materials of construction), volatility, costs, and incompatibility with surroundings. A graph of the empirical results, or electrical behavior of such cells, is shown in Figure 6.29. It is typical of simple cells with no provisions made in their design for diffusion limitations, solidification of reagents, or any other performance controls. These early cells illustrate the very basic characteristics one can expect from this class of concentration cells.

Figure 6.29 UU carbon, sybron separator, spec. Grav. soln, +1.2.
These curves were generated by discharging cells across a constant resistive load – not the optimum arrangement for either high efficiency or good performance. However, they do demonstrate the general characteristics of voltage rise and decay during charging and discharging, respectively. One of the main purposes of such extended cell testing is to learn a bit more about life and cycling limitations. Over 5,000 cycles have been accumulated on single cell laboratory devices with no discernable deterioration in performance or chemical composition of the electrolyte or erosion of electrodes, thus making predictions of cycle life rather difficult with no degradation data from which to extrapolate.