
Chapter 8
Polysulfide – Diffusion Analysis
The ensuing analysis was prompted by problems encountered with the sulfur (–) electrode while developing a bromine/sulfide secondary cell for load leveling application possibilities. The lives of our composite carbon/plastic, negative electrodes were severely limited upon extensive cycling. After much research and experimentation, the puzzle of limited electrode cycle life was solved. The problem was caused by the erosion of carbon particles from the composite structure by the formation of hydrogen gas just under the surface of the somewhat porous plate. The polymer (plastic) component adheres very well to the total electrode structure and has a physical continuity that does not characterize the very frangible carbon components. After extended operation of this sort with gasses ejecting carbon, the electrode acquires a very high interface resistance and ceases operating as a functioning electrode, producing mono-sulfides due to starvation in the depths of the holes left by the separated carbon.
It is most important that we try to take into consideration any and all possible processes and mechanisms in our analysis of the experimental observations. It must also be understood that much of what is offered as explanations for cell behavior is in the realm of speculation – to be verified as we continue with these empirical studies.
The dynamics of molecular diffusion, ionic conduction, and the oxidation-reduction processes that occur at the electrode/electrolyte interfaces are considered here with a view toward simplifying and qualifying. In concentration cells, these are considerations of prime importance in the study of the balance of materials transport. Certain operational models must be assumed for such an analysis to be initiated and progress. As we learn more and attempt to match the theoretical modeling to empirical data, some of these assumptions may be abandoned or changed in order to provide a better picture of what actually occurs within a cell.
In concentration cells, in which the situation usually involves the movement of uncharged molecules as well as ionic species toward and away from electrodes necessary to its operation, a great amount of attention must be devoted to these transport mechanisms (Table 8.1). In the pages to follow, a straightforward examination of the ionic and molecular transport processes that occur within an electrochemical cell are examined. The subject cell makes use of sulfur/polysulfide reactions at electrode surfaces. It is the intention that this simple exploration of the diffusion balances will enable us to better assess cell performance and charge capacity for not only the sulfide cell but for other concentration cell systems based on different chemistry.
Table 8.1 The transport of various species.
| Transport mechanism | Toward electrode | Away from electrode |
| Electric field assist | Na+ | S= |
| Neutral diffusion | H2O | H2 |
| Electric field retard | SxS= |
The sections to follow discuss the various mechanisms that take place within electrochemical cells of this type.
Various mechanisms take place within electrochemical cells of this type and present a preliminary analysis of current density distribution and reagent diffusion at the surface of porous electrodes. This initial review is intended primarily to provide a basis for some further attempts at characterizing the kinetics that occur at the surface of the negative electrode.
8.1 Polarization Voltages and Thermodynamics
Voltage losses within a cell, other than those encountered by the electrolyte ionic resistance and electrode electronic resistance, can be classified into three categories:
- Interface resistance, or ohmic over-potential is due to the resistance of “contact” between electrolyte and electrode.
- Activation energy, or the rate with which a reaction proceeds, is associated with an energy barrier or activation over-voltage.
- Concentration over-potential is a polarization voltage that is a function of the concentrations of reactants and reaction products.
These factors may be derived and represented as follows. The Nernst equation for an electrode reaction of the form
(8.1)
is given in general terms as
(8.2)

where R is the universal gas constant, F is the Faraday number, and n is the charge on the ion.
W is defined as

where the a-terms are the activities for the reactants and the reaction products.
For the reaction of interest at the (–) electrode during the charge mode, (see equation (8.5)), equation (8.3) becomes
(8.4)

Since the activities depend on concentration, or population density of the reactants, high over-voltages can be generated under severe starvation conditions at electrode surfaces. This possibility is especially real if cells are charged by constant current sources. Some cell operating conditions can lead to the evolution of hydrogen, thus tending to further complicate conditions and contribute to additional starvation of electrode sites.
Our primary interest at this time is in the mechanisms associated with the negative (sulfur/sulfide) electrode and, more specifically, the processes involved in the charging mode. Since precisely the opposite occurs at the positive electrode during “charging” in a symmetrical concentration cell, the reverse processes can be analyzed to provide complete cell analysis.
8.2 Diffusion and Transport Processes at the (–) Electrode Surface
The processes and reactions that occur at the (–) electrode during charge are more complex and “limiting” than those during the discharge mode. The solubilized sulfur attached to S= ions are supplied only by mechanical means or thermal diffusion against the (–) electrode repulsion. Sodium ions, on the other hand, migrate toward the (–) electrode via electric field attraction.
Figure 8.1 shows the transport processes of interest as defined by us for further examination.

Figure 8.1 Negative electrode in charging mode.
For the primary energy storing reaction, shown below, to take place at the (–) electrode,

There must be adequate supply of not only Na+ ions to the surface, as demanded by the charging current density, but also a corresponding quantity of polysulfide. If these conditions are not met, then starvation at the electrode surface will occur, and if the voltages are high enough, water will be broken down and hydrogen will evolve in accordance with
(8.6)

As shown in Figure 8.1, if some hydrogen is produced, then it further complicates the situation since it too must be removed from the reaction site to enable the reduction of sulfur to take place. To summarize, the transport of various species may be put into the following categories.
There is the obvious necessity to provide high accessibility to the (–) electrode, especially during the charging mode since only the sodium and mono-sulfide ions are assisted by the electric field gradients and the polysulfide must be provided at the sites by forced flow for higher current densities.
8.3 Electrode Surface Properties, Holes, and Pores
In order to enhance the electric current density of the frontal area of the electrode, it is quite reasonable to think in terms of increasing the total available working area by making the electrode surface porous or in terms of providing a high degree of irregularities on the surface, instead of a smooth, dense, non-porous electrode. As this direction is taken, the electrode area is increased, and the effective, or working, electric current density for any given frontal area current density is decreased.
This would seem to be better since the demand rate for reagents to and from the electrode (micro-surfaces) is reduced, thus lessening the chances for starvation and high polarization potentials to appear. However, one must consider the fact that this increased surface area is not as readily available to the reagents as the smooth frontal area was, and the electric field strength within valleys and pores will be diminished.
The following is a simple exercise as a very preliminary attempt to explore the interrelations between increased electrode “porosity” and its effect on performance.
As a first step in the investigation, the shapes and intensities of electric fields and field gradients were examined as functions of geometry. Figure 8.2 shows some of the more direct and early approaches toward increasing electrode area per unit frontal (normal projection) of a flat plate electrode.

Figure 8.2 Shaped electrode fields.
As can be seen in both cases above, the working surface area has been increased by either putting a portion of the electrode at an angle or by making an indentation in the electrode surface.
Looking at the electric field, one sees that the field gradient is also changed, and the electric current will be modified accordingly as well. The question here is whether there is a net gain in performance and how much.
Much has been done in the past regarding electric field configurations for various geometrical shapes of conductors. One excellent source is W. R. Smythe’s Static and Dynamic Electricity published in 1950 by McGraw-Hill Co. Unfortunately, even the simplest of geometry leads quickly to very lengthy and complex computations for field shapes.
Consequently, we have taken a much more simplified approach merely in order to see trends in the results of providing pores and holes on the surface of electrodes.
Smythe (see Bibliography reference 26, particularly Chapters I and V) and others have explored the solution of Laplace’s equation for plane conductors with infinite extension, or slotted planes. For example, the application of the Schwarz transformation to the computation of charge distribution, σ, in the x-direction on a plane conductor with a slot of width 2a gives the following expression:
(8.7)

The conductive plane extends infinitely in the x and y directions, and the slot of width 2a extends indefinitely in the y-direction. In this case, an electric field exists in the z-direction, imposed by parallel plate conductors extending in the x and y-directions.
An approach we have taken here to approximate the field strength within a hole or slot, as compared to the electric field intensity at the plate surface, is as follows.
Figures 8.3a and 8.3b show configurations of parallel plate conductors as a means of establishing extreme conditions of geometry. The first drawing is for a conductor that has a very large area (slot width). In this case, the electric field is quite uniform within the slot, and the voltage linearly varies with the distance in the y-direction. In other words, the field strength, or voltage gradient, φ, is
(8.8)


Figure 8.3 (a) Electrodes with large area depressions. (b) Electrodes with narrow slots.
within the slot and E/L at the plate surface, whereas the field strength is close to zero in the very small slot shown in the second drawing. The electric field does not bend very much into the narrow hole. The field for this case is approximated by
(8.9)

both within the hole as well as at the surface of the plate.
In essence, for deep and very narrow slots or holes there is no electric field within the depression.
Now consider the situation where the size of the slot is in between the two above extremes, as shown in Figure 8.4.

Figure 8.4 Dimensioned hole or slot.
In this case, which more closely represents actuality, the electric potential drop, ∆E, from the top of the hole to the bottom can be expressed in numerous approximate ways. One very simple view is to let the portion, ∆E, which appears in the pore of the total field, E, between conductors, be further modified by a parameter, β, as follows:

where β is a multiplier or an adjustment factor dependent on the ratio of depth to width of the slot or hole. Perhaps, a workable representation of the manner in which the field strength varies within a pore in accordance with the ratio of w to λ. is
(8.11)

An equally useable expression could be

where α is a constant of proportionality.
In both cases, equations (8.11) or (8.12), β goes to zero as w/λ, and β goes to unity as w/λ becomes large.
For small pores, equation (8.12) can be approximated by β = (w/λ) α, and then the complete expression, equation (8.10), for small pores becomes
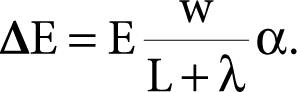
Assuming a linear field, the voltage gradient within the hole is

The approximation is not valid in those cases where the pores, or electrode surface depressions, are shallow and have large areas. Since these are not the conditions of interest to us, and the mathematics are unnecessarily more complex to cover the entire range, this approach has been taken for the present.
If the above is a reasonable first approximation for the ion attractive force producing fields within electrode pores or depressions, then the next step is to examine the magnitude of electric current densities that result from such surface irregularities.
8.4 Electric (Ionic) Current Density Estimates
Ionic mobility, µ, in the y-direction is given as µ = (y/t) (1/φ), where t is time an φ is the electric field gradient, or φ = ∂E/∂y, and the ionic speed is
(8.15)

More useful to this analysis is the specific resistivity, ρ, of the electrolyte, expressed as
(8.16)

where
(8.17)

and A= area normal to the current flow.
By applying equation (8.13) and solving for the current, i, in the pore entrance where the electric field gradient, φλ, is as described in equation (8.12), we obtain
(8.18)

Since the expressions are all normalized for a unit thickness of electrode into the plane of the page (the z-direction), area, A, is equal to w, and equation (8.14) becomes
(8.19)

This can be compared to the current of Ew/Lρ at the top surface of the plane conductor. It is seen that, as the width of the slot is made smaller and the depth, λ, is made larger, the current into the pore decreases, and the current densities, i/w, also change in accordance with
(8.20)

into the pore, and
(8.21)

at the top surface.
As the electrode becomes more porous, i.e., more internal surface area per unit frontal area of the plane face, the current density will reduce proportionately. However, if we further assume, as another first approximation, that the current density within the hole is uniformly distributed along the entire available surface area of walls and bottom, then the expression for the current density, σp, within the pore becomes
(8.22)

It should be also remembered that the specific resistivity, ρ, of the electrolyte within the pores will not be constant as the depletion of charge carriers continues with charging. When one integrates the total current across the face on the porous plate electrode, it also becomes apparent that the largest portion of the current flow is to the outermost surface of the electrode and not within the pores.
8.5 Diffusion and Supply of Reagents
The supply of free sulfur to the (–) electrode takes place only by molecular or thermal diffusion or forced convection. Since attachments to negatively charged sulfide ions solubilize the free sulfur, the difficulty of supply is compounded by the electric repulsion of the (–) electrode. Perhaps, the greatest problem associated with over potentials arises from the difficulty of sulfur supply during charge. If we assume a uniform field gradient into a pore, then, in order to maintain a steady rate of supply, (dQ/dt)S, of SxS= during charge into the pore, the rate must be equal to the charging current into the pore with entrance area w, or
(8.23)

Figure 8.5 represents a pore with concentrations Ca and Cb outside and inside, respectively, of the pore.

Figure 8.5 Carbon conductor electrode pore.
In accordance with Fick’s First Law, the rate of diffusion is directly proportional to the concentration gradient or difference across a boundary:
(8.24)

Applying the relationship above,
(8.25)

where R = Cb/Ca.
In reality, the diffusion situation for the supply of SxS= is worse than described above. Since there is in fact concentration gradient all along the pore length, λ, the availability of sulfur diminishes at greater pore depth.
The question that arises now is whether the value of R must be so small that, to maintain the balance of sulfur flow into the pore to match the conversion rate of sulfur into sulfide ions, severe polarization will be encountered and hydrogen gas will be produced.
Under the best of conditions, and from equations (8.16) and (8.21), the rate match is

and that does not account for the electric repulsion forces of the (–) electrode. Solving for R in equation (8.26), we obtain
(8.27)

Since no materials are lost (plated on) or generated (plated off) within the pores, only conversion takes place, and spent products must be removed and active reagents must be provided. Mono-sulfide is removed from the pore by diffusion processes but assisted by electric fields, whereas the same fields retard the supply of polysulfide.
Composite expressions can be devised in order to describe the total migration rates of the various species by introducing some additional factors associated with coulombic forces.
8.6 Cell Dynamics
8.6.1 Electrode Processes Analyses
The general expression describing the half-cell potential at the (–) electrode for the charging reaction, S + 2e– → S=, as given by the Debeye-Huckel theory and the Nernst equation, is shown in equations (8.1) through (8.4).
We will proceed to outline an initial approach to solving the problem or answering the questions that surround the shapes of the charging voltage curves for an LS-2 cell.
8.6.2 Polymeric Number Change
It should be noted that the sulfur/sulfide concentration cell has an alkaline electrolyte due, in part, to the hydrolysis of the Na2S salt in accordance with the following reaction:
(8.28)

If a salt, AB, is dissolved in water, it will undergo some degree of hydrolysis. This can be expressed simply as
(8.29)

The extent to which this occurs is usually measured as the degree of hydrolysis. In the case of a salt of a weak acid and strong base, as in this instance, the equilibrium constant, Kh, for hydrolysis may be expressed as
(8.30)

The ionization equilibrium constant, K, is generally expressed as
(8.31)

The ionization constant for NaOH is orders of magnitude greater than that of H2S, hence the resultant solution is quite basic.
In the sulfide/polysulfide alkaline concentration cell, the important issues of the behavior of sulfide ion and sulfur atoms in the immediate vicinity of the electrodes become quite important. These concerns exist during the charging and discharging processes, as well. The situation is different than what is normally encountered in electrochemical cells because of the fact that both the oxidizing agent, S, and the reducing agent, S–2, are the same chemical specie. Furthermore, the output potential is not derived from a difference in chemical potential between two different species, but instead is a consequence of differences in concentrations of the same chemical element.
To review, again, the elemental sulfur, S, is the oxidizer when being reduced by the addition of two electrons from an external voltage supply circuit at the negative electrode during charging. The exact opposite takes place at the electrode during the discharging mode when electrons are given up to the negative electrode and then subsequently conducted to an outside electric circuit load.
We will now take a deeper look into the current density distribution and reagent diffusion at the surface of porous electrodes. This initial review is intended primarily to provide a basis for some further attempts at characterizing the kinetic actions that take place at the surface of the negative electrode in a sulfide electrolyte. This analysis is independent of the processes that occur at the opposite, positive electrode because the electrolytes are assumed to be independent. The only transfer between electrolytes across a cation membrane in this analysis is presumed to be Na+, sodium ions.
The initial form of the electrolyte when the cell is in the discharged state is presumed to be Na2S5, or the penta-sulfide form, which can be represented as Na2S · S4. There are then four sulfur atoms available as essentially free sulfur attached to the sodium mono-sulfide in solution.
If we now consider the various sequences in which the sulfur becomes detached from the salt during the charging processes, the following seem to be the available choices.
- Random generation of various polysulfides, Na2S · Sx, where x can have values from 0 to 4 – In this case, we would assume some statistical distribution function to describe the molecules in solution at any point in time.
- To assume that the SxS= decreases in size by one sulfur atom at a time during charge and then promptly leaves the immediate electrode surface region to make way for the next SxS= ion until all SxS= species have been converted to Sx–1S= – The next stage in the reduction of size would take place until, via such systematic procedures, the electrolyte is converted to Na2S at full charge.
- To assume that each SxS= goes all the way to S= in a series of consecutive steps and then leaves the electrode surface area – This assumption would state that there is always only a ratio of S= to S4S= with which to contend.
In any event, if we let the availability of sulfur for reduction to sulfide be the same independent of the multiple attachment factor to sulfide, there should be little practical or analytical concern here as to the exact distribution of sulfur at any time during the charging process.
Figure 8.6 shows the diffusion rates in which we are interested for this present analysis. The drawing diagrams the simple but essential aspects of a half-cell. The volume, V, indicated in the figure includes the entire negative electrolyte volume for a unit area of (–) electrode, including that which is stored in the reservoirs external to the cell. Hence, the distance between the membrane and the electrode is not representative of the true geometry.

Figure 8.6 Diffusion representation.
We will use method #3 above to represent a species in the analysis regarding the availability of S (or concentration of S) as independent of x, or the total concentration of S in the form of SxS= taken as |S|. Now we have only two kinds of anionic species in solution in the negative electrolyte of volume V, (other than OH– and any Br– ions that have migrated through from the (+) side), and they are S= and S4S=. Then, the diffusion processes to and from the electrode surface can be represented as shown in Figure 8.7. It should be noted that there is no storage (build up or depletion of reagents at the electrode surface) of reagents within some region of immediate proximity to the electrode surface.

Figure 8.7 Diffusion to and from electrodes.
There are, in fact, six analytical approaches that we will try to take toward describing the kinetics at the electrode surface. These will be taken in succeeding steps and are listed below.
- Case 1 – Flat surface electrode, no surface storage region, instantaneous and perfect mixing of reagents throughout the volume, V, no hydrogen generation
- Case 2 – Flat surface electrode, some reagent storage at the surface, instantaneous mixing of reagents within each of the two volumes, volume v is designated as the surface region storage, volume V is designated as the bulk (–) electrolyte, no hydrogen generation
- Case 3 – Porous Surface electrode, some storage region, instantaneous mixing in both volumes, no hydrogen generation, smaller diffusion coefficient
- Case 4 – Flat surface, some storage, some hydrogen generation, instantaneous mixing
- Case 5 – Porous surface, some storage, some hydrogen generation, instantaneous mixing
- Case 6 – Porous surface, some storage, some hydrogen generation, reagent concentration gradient at electrode surface volume, instantaneous mixing within volume V.
For perfect, instantaneous mixing there are no diffusion equations. Only the concentration changes as the various species are generated electrochemically. Referring to Figure 8.7, we see that the expression describing the half cell potential, E, is
(8.32)

and it is the final expression for this particular, somewhat unrealistic case of full access to the electrode surface, with no stagnant electrolyte region at the electrode surface.
Activity coefficients for sulfide and sulfur are being researched in the chemical literature. Meanwhile, since we are primarily concerned here with functional relationships, we can proceed with a general analysis of the electrode processes. At present, our main interest is in the functional dependency of, and trends in, the voltages during cell operation.
Figure 8.8 gives a few values of generally available activity coefficients for various compounds in aqueous solution (Latimer 1952) where γi = activity coefficient for the sulfide ions = aS–/|S=|, and γs = activity coefficient for the “free sulfur” = asx/|S|.

Figure 8.8 Activity coefficients for strong electrolytes from Latimer’s oxidation potentials.
The concentrations of the species are further defined in this particular case as,

and

There is also a useable relationship between Qi and Qs. The total of the numbers of sulfide ions and sulfur atoms remains constant since one sulfide ion is produced for each sulfur atom lost during charging.
Qi + Qs = Qo equals the sum of both species present initially, and Qs = 4Qi.
At any time, t, later the expression assumes the form
If we normalize the analysis for a unit of electrode area, then the volume in the main negative solution per unit area is V.
Now it is necessary to address the rate, Rg, of sulfide ion generation during charge. This can be expressed as
(8.36)
where α = constant of proportionality, σ = current density, amps/ unit area, and η = coulombic efficiency.
Thus, we may represent the rate of generation, dqi/dt, of S= ions as

which is also the exact rate of reduction of available sulfur.
Since Qo + 4Qi = Qi by substitution between equations (8.37) and (8.32), we obtain

Solving for qi by integrating equation (8.33),
(8.39)
From the limits of the problem when t = 0,
(8.40)

and the final expression becomes
(8.41)

It is now possible to place the values for qi and qs into equation (8.26) to solve for E as a function of time.
Using the relationships in equations (8.27), (8.28) and (8.29), and substituting into equation (8.26),
(8.42)

Substituting the value of qi from equation (8.33) into equation (8.34),
(8.43)

is the final expression for this case of full access to the electrode surface and no stagnant region at the electrode surface.
We have not yet found the activity coefficients for sulfide and sulfur in the literature. However, since we are primarily concerned here with functional relationships, the details of specific values for the activities can be left for a later time when we become interested in more dependable, quantitative evaluations of these expressions to compare with experimental results obtained in the laboratory.
At present, our main interest is in the functional dependency of, and trends in, the voltages during cell operation. It appears that the chemical potentials of neutral and S= ions and neutral S are independent of whether they are “polymerized” or not, and that the effective reaction is S + 2e– → S=. While this is an approximation, it is quite reasonable given that the half-cell potentials for the reactions given below are so close together.

Since pH is usually very high, reactions of the form HS– → H+ + S= and the presence of HS– ions are ignored for the present.
Figure 8.8 is a plot of a few values of generally available activity coefficients for various compounds in aqueous solution (Latimer 1952).
An interesting graph is the plot of half-cell voltage versus time as expressed in equation (8.37). In order to generate such a plot, the values of a few constants are needed. The values are listed below. Some assumptions or simplifications have been made in a few coefficients outside of the values of the physical constants.
- R = universal gas constant = 1.99 cal deg–1 mole–1 = 8.3 joules deg–1 mole–1
- α = conversion constant = mole/52 amp-hours = 0.0019 mole amp–1 hour–1 = 5.34 × 10–6 moles amp–1 sec–1
- η = coulombic efficiency = 1.0
- γi = γs = as an interim assumption
- V = storage volume in liters per unit area of electrode
- n = number of electrons transferred per event
- Eo = 0.50 volts half cell voltage at standard conditions
- Z = Faradays number = 96,500 coulombs/equiv = 26 AH/equiv.
Now we need to assess the values of σ, Qi, and Qs on the basis of a particular current density. Since most of the experiments performed at TRL with single cells of a 25 in2 area were performed between 4 and 6 amps, we will take 0.03 amps per cm2 as the current density. A total charging time of 5 hours is also commensurate with most of the data accumulated to date.
Thus, a total charge of 0.15AH/cm2 corresponds to the capacity of the electrolyte of volume, V.
Now, there are Qs = 0.15AH/(52AH/mole) = 2.9 × 10–3 moles of available S, and Qi = Qs/4 = 7.2 × 10–4 moles of S=.
Substituting the above values into equation (8.37), the following is obtained:
(8.44)

Figure 8.9 is a plot of equation (8.38) of half-cell voltage versus time in hours. Equation (8.38) then has the form
(8.45)


Figure 8.9 Half-cell potential versus time.
Ohmic resistance of the electrolyte and any interface resistance have not been included in the plot.
As can be seen, the voltage is below 0.5 volts at the beginning of charge when there are relatively few sulfides present, as compared to the available sulfur. As charging progresses, the potential rises fairly linearly until the concentration of sulfides becomes great and that of available sulfur has diminished significantly.
In theory, the cell potential would rise to extremely high values if the charging were continued in the constant current mode. In reality, as the charging potential rises high enough the decomposition of water would take place, and hydrogen would evolve.
The factors influencing the shape of the plot include the following:
- relative values of γi and γs
- the manner in which γi and γs change with or depend on the concentration of the electrolyte
- electric current density
If, for example, the ratio γi/γs were to be greater or less than unity, as was assumed in the exercise above, the general shape of the voltage versus time curve does not change. Rather, it is displaced on the vertical axis accordingly, as shown in Figure 8.10.

Figure 8.10 Half-cell potential versus time.
The data calculated and plotted above assumes that the activity coefficient ratios are constant throughout the range of solution concentrations. In reality, they probably do vary between the two extremes of very dilute to very concentrated. Figure 8.11a graphs two sample functions of the ratio of r = γi/γs.

Figure 8.11 (a) Activity coefficient ratios versus charge state. (b) Half potential versus time.
For the low-mid values, the ratio r1 = γi/γs starts at 1.5 and goes down to 0.5 midway, and then it returns to 1.5 at the end of charge. In the second instance, r2 = γi/γs starts at 0.5 and goes up through values of 1.5 and returns to 0.5 at the end of charge.
The purpose is to observe the effect of widely varying ratios, r, upon the charging curve for the half-cell.
Figure 8.11b gives plots of the cell voltage for the two r-functions above.
The shapes change noticeably with such a spread of values, one with a peak in the mid-concentration range and the other with a saddle in mid-range.
The preceding analysis was performed for the very simplified situation, Case 1, where there is not a boundary layer on the electrode and the idealized perfect mixing of electrolyte components.
The analysis continues with the circumstances described in Case 2. This next stage in the development of descriptive modeling more closely approximates actual conditions within a cell. It is expected that these exercises will significantly improve our investigations for electrode structures with lower polarization losses and will explain the seemingly peculiar voltage versus time curves and their changing behavior with cycling.
A graph would plot the half-cell voltage versus time, as expressed in equation (8.35). In order to do so, the values of a few constants are needed. They are set as the following:
- R = universal gas constant = 1.99 cal deg.–1 mole–1
- 8.3 joules deg.–1 mole–1
- α = conversion constant = mole/52 amp-hours = 0.0019 mole amp–1 hr–1 = 5.34 × 10-6 moles amp–1 sec–1
- η = coulombic efficiency = 1.0
- γi/γs = as an interim assumption
- V = storage volume in liters per unit area of electrode
- n = electrical charge per ion = 2 equiv./mole
- Z = Faradays number = 96,500 coulombs/equiv. = 26 AH/equiv.
By substituting the above values into equation (8.35), the voltage values of half-cell potentials can be obtained.
8.7 Further Analysis of Electrode Behavior
8.7.1 Flat Electrode with Some Storage Properties
The earlier analysis assumed simplified conditions, in which no delayed boundary region existed at the electrode surface for reagent storage and instantaneous solvent mixing.
The purpose of this analysis and report is to present a sequence of studies aimed at understanding the fundamental processes associated with cell behavior at the negative electrode, in particular. The bromine electrode is comparatively well behaved and presents no inexplicable or unwanted electrical characteristics.
The conditions under which this next stage in the analysis has been done are described fully in the text. The results are realistic and provide valuable assistance in understanding the rate process balances.
A constant current mode of charging was assumed, and the coulombic efficiency is set at 100% all the way through. In reality, the current efficiency very much depends on current density and reagent concentrations. In the next sequence, we will explore cell behavior when current efficiency is treated as a function of both current density and polysulfide concentration in the immediate vicinity of the (–) electrode.
The conditions we will examine are as follows:
- Case 2 – Flat surface electrode, some reagent storage at the surface, instantaneous mixing of reagents within each of the two volumes, volume, v, is designated as the surface region storage, volume, V, is the bulk (–) electrolyte, no hydrogen generation.
Figure 8.12 illustrates the regions in question and the distribution of reagents at any time within these two regions.

Figure 8.12 Flat electrode with two associated storage regions.
The concentrations of sulfur and sulfide are considered uniform at all times throughout the respective regions. In other words, the concentration of SxS= for each value of x is assumed to be uniform in volume, v, and volume, V, separately. The chart in Figure 8.12 illustrates this idea that the concentration of each species is constant at all times in the cell regions.
Figure 8.13 depicts the rate processes for the gain and loss of sulfide ions and available sulfur for each of the two cell regions.

Figure 8.13 The rates of gain and loss of sulfide ions.
Sulfide ions and available sulfur are entering and leaving the regions across the boundary (artificial separation of the two regions) by diffusion. Also, sulfide ions are generated at the rate Rg(–) into the boundary layer at the electrode surface by electrolysis (reduction). Available sulfur is lost at the rate Rg(S) to the region by the very same electrolysis process. Hence,
(8.46)

is the generation rate.
Figure 8.14 identifies the various species within the solutions. In order to minimize the complexity of the symbols, the ionic species in each of the two regions have been given the following designations for the amount of reagents present at any instant of time during the charging process:
- qib = quantity of S= ions in the boundary layer at the electrode surface
- qsb = quantity of free sulfur within the electrode surface boundary layer
- qir = quantity of sulfide ions present in the reservoir region
- qsr = quantity of free sulfur in the reservoir region.

Figure 8.14 Identification of ionic specie regions.
The rate, Rg(–), with which sulfide ions are being generated by the charging process is
(8.47)

and the rate with which free sulfur is reduced is
(8.48)

The net rate, dqir/dt, of increase in sulfide ions in the reservoir region (volume, V) due to sulfide ions leaving and entering the boundary region via diffusion of both monosulfides and polysulfides into and from the reservoir is represented as
(8.49)

The rate, dqsr/dt, of increase in free sulfur in the reservoir region due to diffusion into and from the boundary is
(8.50)

where λ is a diffusion path length.
In volume, v, or the boundary region, the rate balance equations are

and

The diffusion constants represented by k for the sodium mono-sulfide and the sodium penta-sulfide molecules should be self-explanatory. There are four diffusion terms in the expression for dqib/dt above because available sulfide ions are transported via both the polysulfides and the mono-sulfides.
Since immediate mixing in each of the regions is assumed to take place, no provision has been made here to account for any repulsive electrical forces of the (–) electrode acting upon the different ionized components. Hence, in this treatment any deficiency or surplus of ionic species at the immediate surface of the electrode is accounted for in the layer referred to as the boundary region.
Since we are concerned at this time only with the neutral molecular diffusion of the reagents, and not ionized charge layers, the diffusion constant, km, and kp, for Na2S, and Na2S5 respectively, are considered the same whether they are in the boundary region diffusing away from or toward the (–) electrode or in the reservoir region drifting in either direction.
Now it is possible to solve for a set of simultaneous differential equations that describe the concentration balances at any time during charging.
There are four variables and four equations:
Since volume, v, is so much smaller than V,
(8.54)
The variables that are available in this analysis are v and k.
Proceeding with the mathematical operations, the following ensues. For purposes of convenience of representation, let

To simplify the use of symbols for representing the various constants and multiplier factors in the equations, this interim shorthand notation is employed:
(8.56)

(8.57)

(8.58)

(8.59)

(8.60)

Representing the differential operator with the symbol D in equations (8.49) through (8.52), we have a series of relationships that assumes the following forms:

We are particularly interested in solving for the terms qib and qsb the quantities, respectively, of sulfide ions and free sulfur within the boundary layer at the (–) electrode.
Equations (8.49a) and (8.51a) contain too many q terms for easy solution. Hence, in order to solve for the variable, qib, the quantity of sulfide ions and free sulfur in the stagnant, or boundary, layer region we must first solve for qir, and qsr by equations (8.50a) and (8.52a). Using these as selected simultaneous differential relationships to be solved, the following are the manipulations.
Taking equations (8.50a) and (8.52a) we have,

Since the model assumes perfect mixing in the reservoir region, and since the distance to be traveled by any diffusing specie can be from the outer edge of the boundary region or from within the region, the average distance to be traveled by that specie is half the thickness of the boundary region.
Thus, we may set the terms λp and λm as equal distances. Then the equations become somewhat simplified because kmr = kmb = km, and kpr = kpb =kp.
Equation (8.52) is multiplied by (D+b), and equation (8.53) is multiplied by bkp, and both equations are added together. The resultant equation gives a solution for qsb:
(8.61)

(8.62)

As a further shorthand notation for simplifying the equation symbols, we set
(8.63)

By adding (8.55) to (8.56), we obtain
(8.64)

or
(8.65)

or
(8.66)

Employing the method for the general solutions to the linear differential equation, we have
(8.67)

where u = complimentary function, and v = the particular integral.
Substituting v into equation (8.55),
(8.68)

Thus,

There are two boundary or limiting conditions from which we may evaluate the constants c1, and c2. They are when t = 0, qsb (t = 0) = Qs = c1 + c2, and the rate processes at the beginning of charge are such that when t = 0,
(8.70)

where the initial conditions within the cell are

Thus, the net rate of production or removal of polysulfides at the beginning of charge, as a function not only of the electrolysis rate but also due to the initial differences in concentrations of reagents within the two regions when t = 0, is
(8.72)

Differentiating equation (8.69) and setting it equal to equation (8.71), we find
(8.73)

or
(8.74)

and

Since c1 = Qi – c2, the final expression for qsb becomes
(8.76)

Equation (8.75) is the first of the two needed expressions for concentrations to be used in the Nernst equation.
8.8 Assessing the Values of Reagent Concentrations
The initial quantity, Qsb, of sulfur in the boundary region is merely that fraction of the total in both volumes, or
(8.77)

Capital Q indicates initial values of solutes.
Similarly, for the other initial quantities as they will appear in the ensuing equations,
(8.78)

(8.79)

and,
(8.80)

The initial quantities in the (–) compartment (negative electrolyte) are related as
(8.81)
where the quantity of ions, Qi, includes the quantity of sulfide ions contained in the polysulfides as well as those in the mono-sulfides. Since there is a one-to-one trade off between ions formed during charging and sulfurs lost, the quantity, Qo, is constant throughout cell operation.
If the molarity of the pentasulfide solution at the start of charge is Mp and the total volume of electrolyte in the negative chamber is v+V, then the number of moles of initial sulfide is (v+V)Mp and 4(v+V)Mp for the available sulfur. Thus,
(8.82)

8.9 Solving the Differential Equations
Now, we must also solve for qsr so that these may be substituted into equations (8.49) and (8.51) to obtain expressions for the required terms qib, and qsb.
Just as we know at any point in time what the relationship is between qsr, qsb and Qo, (see equation (8.71)), we know also that
(8.83)

for the constant rate of electrolysis and concentrated pentasulfide initial solution.
Referring to equation (8.51), the above is substituted for the polysulfide terms so that we may find a solution for qib:
(8.84)

Substitution of terms gives us
(8.85)

Converting to operator format gives us

From the previous calculations, qsb has the form
(8.87)

and the two solutions to equation (8.86) are
(8.88)

Substituting v into equation (8.86), the constants may be found separately as follows:
(8.89)

Taking each equation term separately as it relates to constants, multipliers of t, or exponential terms, we find the following for the constants:
(8.90)

(8.91)


The final form of the solution is
(8.93)

The coefficient c4 is found from the limiting condition of when t = 0, and qib has some initial value of Qs, so we have
(8.94)
These expressions can now be placed into a mathematical spreadsheet program, and qsb and qib can be quantitatively evaluated as a function of charging time or state of charge.
It is now appropriate to arrive at values for the constants K, β, v, V, α, σ, η, Qs, Qo, v, and V.
Diffusion coefficients, D, for various inorganic compounds in aqueous solution at a concentration of 1 molar are given in Table 8.2.
Table 8.2 Diffusion coefficients for various inorganic compounds.
| Molecule | Diffusion coefficient, D, cm2 sec–1 (× 10–5) |
| HCl | 3.44 |
| HBr | 3.87 |
| NaCl | 1.48 |
| KCl | 1.89 |
| CaCl2 | 1.20 |
| KI | 2.06 |
| NaI | 1.66 |
The diffusion coefficient for mono-sulfide and polysulfide molecules in relatively concentrated aqueous solutions can be estimated by employing a spherical model for molecular shapes, Fick’s law, and Stokes’s law.
The resultant formula for calculating the diffusion coefficient is
(8.95)

where the constants, and units are as
- k = diffusion coefficient, cm2 sec–1
- N = Avogadro number, 6 × 1023
- R = universal gas constant, 8.3 × 10–7 erg deg–1 mole–1
- T = absolute Kelvin, deg
- M = molecular weight, gm
 = partial specific volume, cm3gm–1
= partial specific volume, cm3gm–1- η = 0.00894 poise = 0.00894 dyne - sec - cm–2
The molecular weight of Na2S is 78, and that of Na2S5 is 206. Specific gravity data of sodium sulfide solutions taken by laboratory measurement at TRL gives the following values:
- 1 molar Na2S solution sp.gr. = 1.054 (sp.gr. means specific gravity)
- 2 molar Na2S5 solution sp.gr. = 1.233
Molecular weights of the anhydrous salts are as follows:
- Na2S = 78.04 gm/mole
- Na2S5 = 206 gm/mole
In 1 liter of 1 Na2S molar solution there are 1054 – 78.04 = 976 grams of H2O. Since the specific gravity of water is 0.997 at 298 deg, there is then 1000 – 979 = 21 ml of 78.04 gm of Na2S present in the solution. Dividing through that gives for the partial specific volume, v, of the mono-sulfide
(8.96)

Similar computations for the polysulfide gives
(8.97)

Returning now to equation (8.92) and substituting for the constants found, we can compute values of km and kp as follows:
(8.98)

By the same formula, the diffusion coefficient for the pentasulfide is found:
(8.99)

Even though the molecular weights and partial volumes differ considerably, they appear under the cube root, and their effect on the diffusion coefficient is not great.
On the basis of Fick’s law, dq/dt = k(dC/dx) cm sec–1, where dC/dx is the concentration gradient normal to the electrode of that specific solution component in the cell.
The value of Qo is determined by the initial molarity of the pentasulfide at the discharged state and the designed charge capacity per unit area of electrode.
We are now able to calculate half-cell potentials. The two expressions for qsb, and qib, as represented respectively in equations (8.77) and (8.87), are evaluated for graphing potential versus time (or state of charge) for different boundary thickness and current densities at the negative electrode:
(8.100)

In order to estimate the volume of the electrolyte needed per square centimeter, it is necessary to fix the initial molarity, current density, and time of charging to 100%. A period of five hours has been a reasonable time to charge a storage system at constant current, and current densities in the range of 0.04 amps/cm2 and an initial concentration of 0.75 molar Na2S5 have been employed during cell testing on this project. These would result in a total electrolyte volume of 1.28 cm3 (ml) per square centimeter of electrode area, a total charge capacity of 0.20 AH, and a total of 9.6 × 10–4 moles of sodium pentasulfide in both regions.
Now, the two volumes, v and V, can be set to different values to observe how they affect the shape of the charge curves. A new parameter, Γ, is established for the sake of convenience:
(8.101)

At the onset of charge the total number of moles of available sulfur in the 1.28 ml volume of the unit area cell is 4 x 0.00096 moles, and there are initially 0.00096 moles of sulfide ions present. The number of moles of reagents in the boundary region at the beginning of charge is as follows:
(8.102)

We must also estimate the terms a, b, and c from the diffusion coefficients, region volumes, and diffusion path lengths. Since this model is based in part on a stagnant boundary region and perfect mixing in the flowing reservoir region, a reasonable first approximation of the diffusion path ascribable to the reagents is as follows.
The average distance that must be traversed by reagents within the boundary region is λb=1.28 Γ/2, and the average distance (assuming perfectly uniform distribution within the flowing reservoir region) that a reagent molecule must travel from the outer edge of the boundary region to the middle is also λr=1.28 Γ/2. The diffusion path lengths are arguable, depending on electrode design and how far into the boundary region electrolysis takes place.
Consider the following: Since,
(8.103)

and since we also have established that λ is the same for all species,
(8.104)

The binomial term (a + b) appears frequently in the expressions, and its value is
(8.105)

These multipliers are then inserted into the relationships for qsb, and qib in order to obtain the final expressions that can be quantitatively evaluated and employed to provide graphs for cell half voltage as the state of charge progresses for different coulombic efficiencies, boundary layer thickness, current densities, and even diffusion coefficients.
To develop graphs for volts versus time at different settings of Γ and σ, the two expressions for q must be inserted into equation (8.26).
The parametric analysis that has been programmed into a Lotus spreadsheet and database provides a convenient means for examining the shapes of charging voltage profiles as different values of gamma sigma are inserted. Some of these typical results are shown in the accompanying graphs (Figures 8.15–8.24).

Figure 8.15 qsb pentasulfide concentration versus SOC with a current density of 0.04 amps/cm2.
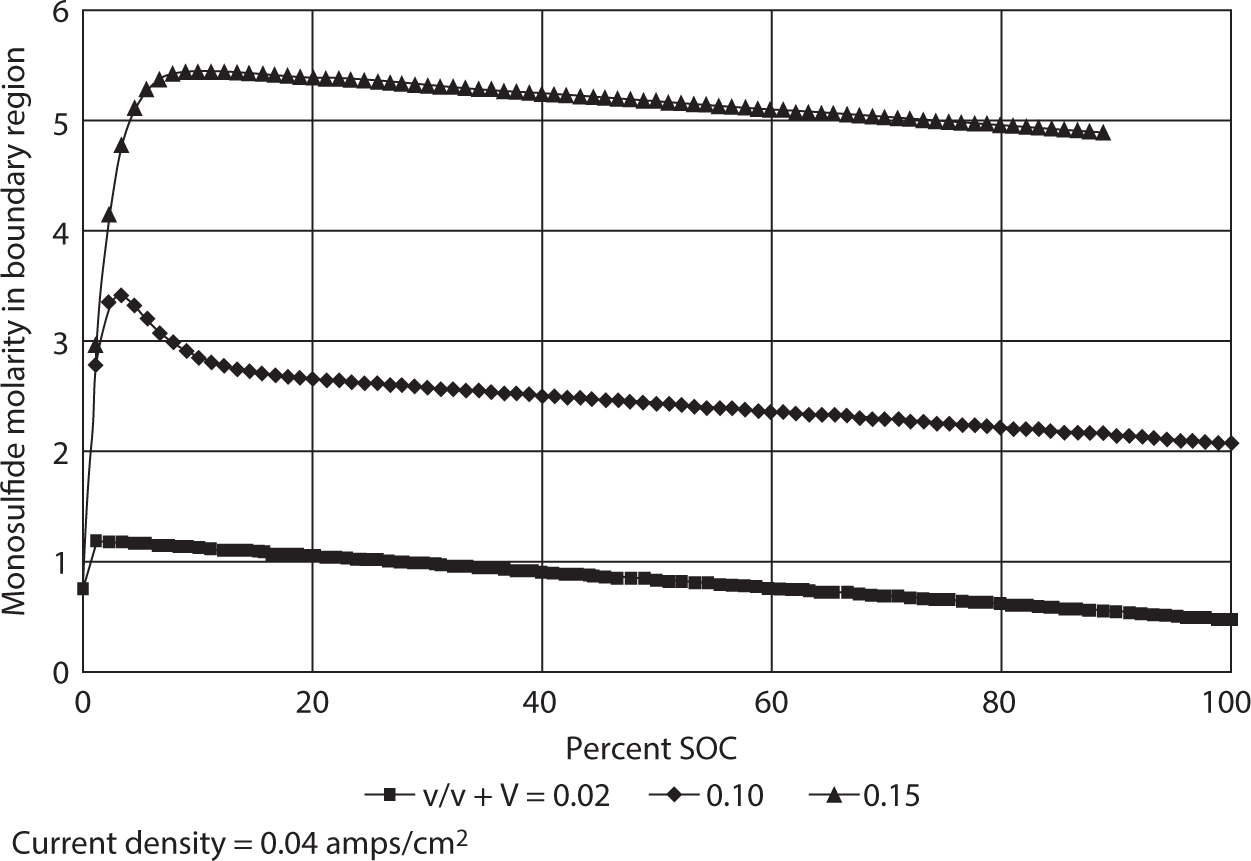
Figure 8.16 qlb monosulfide concentration versus SOC with a current density of 0.04 amps/cm2.

Figure 8.17 ln(qib/qsb) with a current density of 0.04 amps/cm2.

Figure 8.18 qsb pentasulfide concentration versus SOC with a current density of 0.01 amps/cm2.

Figure 8.19 qib monosulfide concentration versus SOC with a current density of 0.01 amps/cm2.

Figure 8.20 ln(qib/qsb) with a current density of 0.01 amps/cm2.

Figure 8.21 qsb pentasulfide concentration versus SOC with a current density of 0.10 amps/cm2.

Figure 8.22 qsb monosulfide concentration versus SOC with a current density of 0.10 amps/cm2.

Figure 8.23 ln(qib/qsb) versus SOC with a current density of 0.10 amps/cm2.

Figure 8.24 ln(qib/qsb) with a current density of 0.10 amps/cm2.
As a first step in the review and presentation, consider how the shape of the volt/time curve depends on the thickness of the boundary region (porosity or accessibility of the electrode surfaces). Extensive experimental data has clearly shown that the smooth, nonporous surfaced electrode gives a flatter shape than a porous electrode with, for example, imbedded active carbon.
It should be noted that, since the mathematics in its present form does not distinguish between positive and negative matter, and especially since the coulombic efficiency is constant, at higher current densities the concentrations could become “negative.” There is obviously no validity to any projected data beyond zero concentrations, and the analysis ceases being useful beyond these limits.
The graphs do clearly indicate that at the higher current densities the limitations imposed upon cell performance, in terms of achieving full charge capacity of the electrolyte, is increasingly limited by thicker boundary layers. The logarithm of the concentration ratio, qib/qsb, is also shown to be dependent upon Γ and σ.
8.10 Cell and Negative Electrode Performance Analysis
These studies are directed at acquiring a better understanding of the mechanisms associated with rate processes at the (–) electrode.
Earlier in this book we presented the beginnings of a parametric study of the diffusion rates and reaction kinetics for the (–) electrode (sulfur/sulfide reaction). This study was prompted by the need for reducing polarization losses and by the desire to explain observed cell performance degradation.
During the past three years of single cell development and testing, we have consistently observed a diminishment in cell performance in terms of voltage efficiency and shape of the charging potential with the accumulation of cycles at high voltage (well over volts). These tests were conducted with two half-cells – one, of course, was the sulfur/sulfide reaction, which is of primary interest to us here, and the other was a predictable and reversible half-cell process such as bromine/bromide reaction. The latter reactions are very well behaved and fairly constant in value.
The sketch, Figure 8.25, describes the essence of our observations. The first cycle of any newly assembled cell has the flat charge shape and slowly sloping discharge shown by curve-A. Initial coulombic efficiency is close to 100%. As cycle numbers increase, the shape assumes that of curve-B. There is a marked rise in charging potential, the discharge curve droops downward, and the coulombic efficiency becomes lower.

Figure 8.25 Deterioration of negative electrode due to H2 evolution.
Significant attention has been paid to the matter of cell performance degradation in the past. Many tests were done in our attempts to isolate the cause of these observations.
Quite obviously, and assuming that the opposite half-cell reaction remains constant over the cycling, there are only three possibilities, which center around the three main factors in cell behavior:
- Electrolytes
- Electrodes
- Membrane
It was determined, via isolation experiments, that changes in neither of the two electrolytes caused the deterioration of performance. After a cell was cycled numerous times and the changes were observed, electrolytes were drained and replaced by fresh solutions. Some return toward initial performance was seen, but even that was only temporary. Cells were taken apart after cycling and membranes were replaced with very little change in performance.
The tests left little doubt that the cumulative changes in cell characteristics were due to electrode changes. If this is the case, the next question naturally concerns the mechanism of electrode property change. There are, once again, only a limited number of speculations possible regarding the source or cause of such changes, which includes the following:
- Sulfur accumulation within the electrode
- Some form of electrode contamination or passivity
- Structural change leading to greater polarization effects.
Of the three possibilities, the third seemed most probable to us based upon much past experience with composite electrodes in salt solution electrolysis.
It has been noted in previous experimental work with electrodes of similar construction that a gradual and inexorable deterioration of its properties occurs when gas is generated at the electrode surface. Based on these experiences, a study (analysis) was conducted to ascertain whether or not a simple mathematical approach to electrode behavior and half-cell potential dependency on diffusion layers bears out our speculations of electrode causes of cell changes.
In the analyses, a dependency of cell potential upon diffusion boundary regions was demonstrated. Since then, the analytical approach has been somewhat simplified, and changes in coulombic efficiency as a function of sulfur concentration have been taken into account.
There are many analytical approaches that can be taken to describe the probable behavior of the ionic and molecular species in the immediate vicinity of the (–) electrode. Some of these are being explored and are discussed in attached separate appendices that will be part of ensuing reports. However, for the sake of expe- diency, and in order to mostly point out the mechanism possibility as an offered explanation for electrode behavior, the following is presented.
In the analysis given for the concentrations of available sulfur and sulfide at the (–) electrode, we assumed a constant coulombic efficiency. In fact, the current efficiency for the reduction of sulfur to sulfide depends at least upon the concentration of available sulfur at the electrode.
A simplified method of initially examining such a dependency was taken, wherein the coulombic efficiency dependency at any point in time during charging is accounted for as a correction factor. In effect, we selected an exponential dependency as follows.
The rate, R, with which sulfur is removed and sulfide is generated is
(8.106)
where
(8.107)
and
(8.108)

A function that satisfies the condition of 100% coulombic efficiency from very high values of sulfur concentration, qsb, (molar range) in the boundary region to very low (0.01 molar levels) is
(8.109)

where C = a constant.
C is evaluated for this exercise by setting η = 0.99 when qsb = 0.01 molar. Then C = 0.001 molar units.
Figure 8.26 shows the manner in which the concentration ratio of, qib/qsb changes with state-of-charge, SOC.

Figure 8.26 Sulfide to sulfur concentration versus SOC.
In all of the calculations and plots, a current density of 0.040 amps/cm2 is assumed since it represents typical values in lab cell tests. The boundary thicknesses of four different values were selected to illustrate the manner in which the slope of the curves depends upon the diffusion layer. As the boundary becomes thicker, qsb approaches small values earlier along the SOC axis.
Figure 8.27 shows the manner in which qib changes with SOC.

Figure 8.27 qib monosulfide concentration versus SOC.
Again, qib arrives at its highest values earlier in the charge mode with increasing boundary value thickness, Γ = v/(v + V) = r.
Naturally, the quotient of qib divided by qsb achieves higher values earlier in the SOC as Γ is made larger. This is shown in Figure 8.28 below.

Figure 8.28 Sulfide to sulfur concentration.
When the logarithm of the ratio qib/qsb is evaluated and plotted as shown in Figure 8.29, it can be seen that the cell voltage will increase higher and earlier in the charge mode as Γ becomes larger.

Figure 8.29 ln (qib/qsb)
According to the Nernst equation, the half-cell potential can be expressed by
(8.110)

Assuming that the activity coefficients remain constant over the entire range of interest, cell potential will increase with the log of the concentration ratios.
It should be noted here that little attempt has been made so far to establish accurate or even reliable approximations to absolute values of constants such as diffusion coefficients, activity coefficients, etc. At this point our main concern is the identification of basic mechanisms for the empirical observations obtained from cell testing.
It is our contention at this point that the major cause of cell charging voltage changes with increasing cycles is due to changesin electrode surface properties. Experience with carbon composite electrodes, where gasses such as hydrogen or oxygen are generated, has consistently shown that deterioration in coulombic efficiency takes place for the generation of other species.
Carbon particles, whether they have the form of activated porous structures, graphite crystals, or very short range “amorphous” carbon, black will be mechanically chipped and eroded away by the formation of occluded gasses.
As these particles leave the eroding surface of the electrodes plastic (Kynar), polyvinylidene fluoride binder is left behind, thus presenting a less conductive outer electrode surface. Ionic species such as polysulfides must diffuse greater distances into the electrode surfaces for electrolytic reduction to take place. The increasing boundary thickness, Γ, tha is synthesized mathematically via the simple modeling above corresponds to this increasing distance for diffusion in an eroding electrode.
Also, as such electrode deterioration takes place, electrical contact (continuity) is gradually lost to any layer of porous carbon bonded to the electrode substrate surface. This loss of contact further reduces the effectiveness of the electrode and even increases the diffusion path beyond that of an eroding substrate.
As the processes continue, the hydrogen will be produced even earlier in the charge mode and in greater percentage, thus further accelerating electrode deterioration.
Some speculations have been made regarding the effects of free sulfur (solid) accumulating within the pores or surfaces of the (–) electrode during the discharge mode. It has been offered that changes in electrode behavior during subsequent charging modes might be due to such factors.
8.11 General Comments
The validity of some of the preceding speculations can be verified or abandoned by further laboratory investigations. It is suggested that these steps be taken to further the study of these mechanisms:
- Test above theory by purposefully placing diffusion barriers immediately adjacent to the (–) electrode to create the conditions predicted by the modeling.
- Examine electrode surface structure after subjecting to severe, repeated cycling.
- Explore solutions to the design of non-eroding electrodes.