
Chapter 7
Thermodynamics of Concentration Cells
Concentration cells are all “redox” in nature because, as is necessarily the case, the reduction and oxidation of chemical species occur within the device for the transference of energy. However, in this instance the redox label does not imply that these devices or cells are full flow electrolyte design with provisions for removing or replacing electrolytes. They are best operated as stationary (static) electrolyte systems. The particulars of design and physical configuration depend upon the intended applications. For example, it may be desirable to use close spacing between electrodes for higher power density application requirement devices, thus proportionately reducing their energy density.
7.1 Thermodynamic Background
The fundamental principles upon which the CIR system is based are probably best identified from the following thermodynamic considerations. Here, we quickly review some of the important and relevant thermodynamic functions upon which the analysis of cell behavior is based.
In order to arrive at the intended expression for the electrical potential of a CIR cell, we will begin with the familiar equation of state for an ideal gas:
(7.1)
where P and V are pressure and volume, respectively, of a quantity of gas consisting of n moles. T is temperature in degrees, absolute Kelvin, and R is the universal gas constant with a value, in the MKS system of units, of 8.31 joules/mole-degree.
To prepare for the relationship for cell potentials, we return to some fundamental concepts. The Gibbs function, G, for any chemical system is given as
(7.2)
where H is the enthalpy, and S is the entropy of the system. Taking the total derivative of G, we obtain
(7.3)
Also, the enthalpy, H, is defined as H = U + PV, where U is the internal energy of a system.
To place the above in more useable terms, we now proceed through the next manipulations aimed at obtaining a final, more useable expression for the free energy (Gibbs function) and the chemical potential.
The total derivative of H is
(7.4)
For neighboring reversible processes,
(7.5)
Substituting for dU in the equation for dH, we get
(7.6)
(7.7)
Substituting the following into the equation for dG,
(7.8)
since T is constant for isothermal processes, the differential free energy becomes
(7.9)
Integrating, we obtain the more familiar expression
(7.10)

The idea of chemical potential, u, is crucial to the study and analysis of electrochemical cells. Here we simply introduce the chemical potential, u, in the following fashion. For a specific constituent, k, of a phase state, uk, is the potential as described by
(7.11)
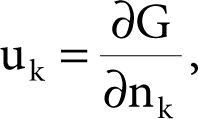
for a multi-component system. If we have but one component, in solution, then
(7.12)
and the chemical potential is the molal Gibbs function and is a function of T and P, or u = G/n, where n = moles.
Now, addressing the electrochemical cell subject, let us convert pressure, P, for an ideal gas into the equivalent parameters for an ionic solution. Additional intermediate terms must be introduced in order to bridge any gap in the continuity of reasoning of thermodynamic equilibrium. The concepts of fugacity, f, and activity, a, have proven to have a significant benefit. If Fm is the partial molal energy and a measure of the escaping tendency, for example, of a vapor from its liquid phase, then the fugacity has been defined by the relationship below:
(7.13)

where B is a constant serving as reference point.
If fo refers to some standard state, then
(7.14)

A more commonly employed parameter in electrolytic solutions is the “activity” of the solution in the immediate vicinity of an electrode. This is usually represented as a = f/fo, and it is usually directly proportional to the molality of dilute solutions because the solute behaves closely to that of a free gas. Hence, we may finally obtain as the free energy, or Gibbs function, of the solution
(7.15)
In determining the voltage, E, associated with chemical potential in electrochemical cells, the free energy may be simply divided by the number of electrical charges that are being transferred in a particular process. In the case of the transference of n moles of singly charged ions at an electrode, the relationship assumes the following form:
(7.16)

where F is Faraday’s number of 96,500 coulombs per equivalent. In general, the equation may be put into the format
(7.17)

where z is the difference in electric charge of the ions in question.
7.2 The CIR Cell
Configurations of the CIR cells may employ two or three of the available oxidation states of an element or compound. A cell is diagrammatically represented in equation 7.18 that makes use of two oxidation states of a metallic element, M. It will be assumed here for illustrative purposes that the oxidation states are M+i and M+j. Then, we may represent a “materially symmetrical” cell employing these agents as

Platinum electrodes are shown in the diagramed cell. They merely represent electrode properties with non-reactivity properties (no chemical participation in the cell processes) and high electronic conduction. In actuality, carbon structures are employed in practical cell designs because of their much lower cost and because the carbon physical properties are better suited to the optimum operation of these cells.
This cell has two chemically inert electrodes, each in its own electrolyte with electrolytes separated by a porous plate. In this case, the separator might be either a microporous membrane that simply retards the diffusion and consequent mixing of the electrolytes, or it might be perm-selective (ion selective membrane). In this instance, the separator could be a cation membrane that permits (+) charged ions such as H+ to pass.
The electrolyte is a solution consisting mainly of salts of the metal, M, and usually a simple acid. Charge transfer within such a cation type cell is principally via hydrogen ions. Very high cell conductivity can be achieved.
The activities associated with each of the ionic species as shown in equations 7.19 and 7.20 are as follows:
On the left side the activity
(7.19)

On the right side the activity
(7.20)

When substituting the activities in the Nernst equation, care must be taken with their order of appearance and algebraic sign of the terms. Perhaps the simplest way to calculate the voltages for a cell is to follow the above format of oxidized form/reduced form. First, we will convert to the log at base 10 for convenience of arithmetic. Then, the expression for voltage will become
(7.21)

Consider the cell described above, in which both sides have identical inert electrodes and iron ions present. In this symmetrical instance Eo is zero.
The voltage for this symmetrical cell is evaluated from the activities of the pertinent ions on either side of the separator for the two electrolytes. The voltage due to the differences in activities for the identified ionic components is found by calculating
(7.22)

Charge, z, is 1 for the ion transport number.
As a result of our cell development the above expression is modified to take into account the preferential storage processes as part of cell design. Electrode processes have been modified to provide for a very selective storage and large activity coefficient enhancement. The design and operational techniques have produced charge storage at cell potentials well over an order of magnitude greater than ascribable to the unmodified Nernst equation. These may be effectively represented in the Nernst type expression as

where α and β are multipliers of the activities due to a dipolemultilayer storage specific to the molecule associated with that ion. During the development of the iron/redox system we have learned how to control the activities of each specific ion in the processes, and there is no metallic iron in its elemental state present in the concentration cell. Hence, it is possible to attain very high electric potential differences in these cells, which makes them practical as energy storage devices. There are no electronically conductive solids at any time present within the cells, other than the inert carbon electrodes. Any solids that might result from cell operating conditions are reversibly soluble and do not result in possible short-circuiting or other deleterious effects.
This approach, with appropriate modifications in design geometry and inactive materials of construction, can be applied to a number of active materials that will dissociate in polar solvents and with two or more soluble oxidation states.
A full flow electrolyte system has some distinct advantages despite its greater complexity and costs. These advantages include (1) the separation of energy density factors from those of power density, (2) an infinitely long charge retention, and (3) total reversibility, in principle.
However, our tests in a limited development program have shown little adaptability of the CIR system to full flow electrolyte configurations. It appears that, in order to benefit from high concentration differences, the reactants are best electro-deposited into porous electrodes that occupy the entire space between the separator and the conductive electrodes. If a cell is designed to permit full flow past the electrode surfaces, then sulfide ions and polysulfides must molecularly diffuse into the depths of the carbon pores. That diffusion process is slow and gives rise to undesirably high concentrations in the bulk electrolyte with consequent diffusion losses through the cell separator. It may be possible with some clever composite designs of electrodes to overcome this deficiency and benefit both from concentration cell performance and the advantages of a full flow system. The mechanical complexities, costs, and size may well be worth it in some applications that have peculiar power and energy density demands or in which long charge retention times are required.
Nature offers few selections from its list of elements and compounds that have all the properties desirable in such redox systems. Among these desirable qualities for practicality are an ambient temperature operation, a low hazard, a plentiful supply of materials, benign behavior, and minimum side reactions that can result in operational failure. TRL has investigated and developed a number of redox-type of systems with varying success in the past. These include the zinc/bromine, iron/redox, and the polysulfide/bromine secondary batteries. Other than the last system, they were all “half redox” cells because only one of the two components of the electrochemical couples are in solution at all times. Both the zinc/bromine and the iron/redox cells suffer from all of the problems associated with the deposition and dissolution of solid metal onto the surface of an electrode. These virtually unsolvable problems include dendrite shorting, non-uniform plating, metal particle fall-off, passivation, and gas generation in acid solutions.
These are very severe problems that have been scrutinized for many years. Unfortunately, these characteristic problems will always be present regardless of how successful some of the cures and improvements in performance become. The polysulfide/bromine system does offer the situation where all reagents are soluble at all times. However, the electrolyte is acidic on one side of the separator and basic on the other. Without some additional subsidiary processes, the pH differences are difficult to maintain. Also, there is the problem of gradual and irreversible diffusion of bromine and sulfide diffusion to opposite sides of a cell as cycling proceeds. TRL has contended with these problems with some degree of success. Unfortunately, these deleterious effects are present and lead to inexorable and ultimate cell failure. Then, it becomes necessary to resort to some complex external method to restore operation.
The most attractive of the above cells, in terms of low cost, safety, and simplicity, is the iron/redox cell, wherein we make use of the three oxidation states, Fe0, Fe++, and Fe+++. The energy storing reaction for the couple Fe0/Fe+++ is
(7.24)
After considerable effort, this couple has essentially been set aside as a practical means of energy storage, not because of its rather limited energy density (maximum of ~75 WH/lb for the dry reagents), but because of its unmanageability with regard to iron plating qualities and hydrogen evolution (hydrolysis and iron attack) in the necessarily low pH electrolyte.
It is possible that a non-aqueous solvent can be employed, in which the iron salts are soluble and have a sufficiently low resistivity so that practical levels of power can be realized from such cells. However, as of this date no significant effort has been expended to search for likely solutions to the problems associated with iron in acidic, aqueous environments.
We have all been searching for a means of electrochemically storing energy that avoids all of the above problems completely and with energy density sufficiently high to make it attractive for large-scale, stationary applications, such as load leveling and solar and wind power. On the basis of our collective past experiences with redox types of systems, we turned our attention to the possibilities of concentration cells. This type of cell has the very attractive property of symmetry. The materials of either side of the cell are the same but at different oxidation states.
An example of a “materially symmetrical” cell is the vanadium redox battery. The vanadium redox cell is not a concentration cell as such. Rather, it is a cell that makes use of an electrochemical couple between three or four of the soluble oxidation states that are available for vanadium. There are problems of reversibility and high costs associated with the materials that tend to limit its application potentials.
The CIR cells rely on only two soluble oxidation states of the same material, or compound, as in iron+2 and iron+3. The potential is easily calculated from the above relationships if we know the activity of the specific ions at the electrode surfaces. Returning to equation 7.23, the Nernst equation, we can calculate voltages from concentrations in dilute solutions where the activities are nearly the same as the concentrations. In order to measure the potential due to concentration differences, one would normally employ a cell with a reference electrode, such as H+/H2, that would provide a known-potential at one end of the cell. However, for the sake of illustration, we can simply postulate a cell with the following conditions.
In an operating concentration cell, we are primarily dependent upon the concentrations of the two primary reactants (oxidation states of iron) at their respective electrodes – in this case, the concentration of Fe++ and Fe+++ at the negative and positive electrodes, respectively. Due to the fact that the sum of all Fe++ and Fe+++ ions on both sides is constant at all times because, for example, as Fe++ ions are generated at one electrode the same number is removed at the opposite electrode, the expression for the voltage in this instance becomes
(7.25)

where φ reflects the interdependence of Fe++ and Fe+++ concentrations in this cell.
It is significant to examine the accuracy of using concentrations in place of activities. Certainly, in very dilute solutions the numerical equivalence of activity with concentration is very close. In fact, for most dissolved materials the values of activity do not change very drastically with low concentrations of the particular chemical species.
The terms c2 and c4 have cancelled, and the concentrations were substituted for activities. Let us look at a magnitude of the voltage one might expect from such a concentration cell. If the concentrations c3 and c1 are 1.0 and 0.01 molar, then the potential, E, would have an impractical, low maximum value of
(7.26)
Proceeding further, it is obvious that a concentration ratio of reactants of 106:1 or more at their respective electrodes would be required to achieve an open circuit potential of 1.0 volts. This is a rather discouraging prediction of the practicality of concentration cells. Our preliminary lab experiments with two or three chemical systems did verify these data.
However, when a charging cycle was performed with a cell with thick porous structure, potentials of over 1.2 volts were obtained. Furthermore, the cells were capable of delivering on a sustained basis a significant electric charge to the external circuit at these levels of voltages. Quite obviously, something else was taking place besides the bulk concentration differences in the electrolytes of a two-compartment cell.
The micro- or nano-porous carbon particles are in intimate, physical, and electrical contact with the electrodes. This type of simple cell with inexpensive components and reagent materials prompted us to explore the mechanisms involved in the storage of charge at such unexpected high values. The same sort of behavior was attained with very similar structures employing compounds of sulfur. The electrical performances of the cells were consistent and repeatable. The sulfur-based cells employ alkali salts of sulfur such as sodium, potassium, and ammonium mono and polysulfides.
The search begins for a plausible explanation of how a cell, with no other energy related processes taking place other than the difference in concentration of the two oxidation states of the same chemical element, can produce such high potentials and sustain high electrical charge densities. If there are no other processes involved, then we must look toward some mechanism whereby the chemical species in question are able to accumulate at the electrode surfaces and give rise to such high voltages.
At present we speculate that the specific ionic species, in this case the ferric and ferrous ions, are injected directly into the pores regions of the microporous carbon at high effective concentrations by the process of electro-sorption. Even though the maximum concentration of the compounds of ferric and ferrous chlorides is limited to not much over 3 molar at room temperature, the population densities of the adsorbed and stored ferrous and ferric substances become equivalent to extremely high activities seen by the electrodes. In order to accomplish this, more energy is required than would normally be to separate the oxidation states during the “charging” process. This condition, for the maintenance of conservation principles, has been observed during cell cycling in terms of the volt/amp inputs and outputs.
There are undoubtedly many processes and mechanisms taking place at the electrode surfaces that call for greater understanding and quantifying. Let us first list and then examine the physical and chemical activity possibilities. Omitting the chances that there are any net or permanent chemical changes occurring in the electrical cycling of the cell, the following are possible, reversible processes:
- Exceeding salt solubility at electrodes during charging, resulting in solid compounds at the surfaces with attendant free energy changes (energy of ionization and dissolution)
- The creation of immense concentration ratios of Fe++ and Fe+++ ions in solution at electrode surfaces
- The adsorption of iron ions and, perhaps, of the salt compounds themselves within the carbon porous structure, which may be Langmuir type processes or Van der Waal’s, depending on whether they are attached as electrically charged components or as neutral molecules with dipole moments.
Meanwhile, the following is an encapsulated glance at the energies of solution, or the dissolution for the ferrous and ferric chloride salts present in the cell being discussed. A perhaps overly simplified, but nevertheless indicative, view is suggested here. Consider the free energies (integral heat of dilution) to infinite dilution as given by Latimer’s “Oxidation Potentials”:
- Fe++ in aqueous solution ~ –20 kcal/mole
- Fe+++ ~ –2.53
- Cl– ~ –31.35
- FeCl2 in solid, crystal form ~ –72.2
- FeCl3 ~ –80.4
If, in the reduction of ferric to ferrous ions at the negative electrode during the charging process, the generation of ferrous salts occurs too rapidly for it to remain in solution, then the process of Fe+++ + e = Fe++ is accompanied by the precipitation of ferrous chloride into the pores of the electrode, i.e.,
(7.27)

Then, there is another exchange of energy involving the heat of solution. Its net value is ∆F = –72 + 20.3 + 2(31.35) = + 7.8 kcal/mole at the positive electrode when charging.
Similarly, one can estimate the free energy at the negative electrode. The net reaction would appear to be
(7.28)

Here, ∆F = –80.4 + 2.53 + 3(31.35) = –16.8 kcal/mole. This result can be related to an electric potential by the relationship below:
(7.29)
where E is in volts, and n is the number of charge changes on the ion.
Upon adding the free energy changes, we see that the voltages associated with this dissolution are in the vicinity of 0.3+ volts, a not insignificant amount. Or, we may represent the processes at the two opposite electrodes during charging as
(7.30)

Free energy for this reduction and dissolution is
(7.31)
and,
(7.32)

where
(7.33)
Also, it is important to examine the dynamics and kinetics of the transport and molecular diffusion.