
6
Metal‐Free Nanoporous Carbons in Photocatalysis
Alicia Gomis‐Berenguer and Conchi O. Ania
CEMHTI CNRS (UPR 3079), Site Haute Température, Université d'Orléans, 1D Av. de la Recherche Scientifique CS 90088, 45071 Orléans Cedex 2, France
6.1 Introduction
After the first works in 1960s and 1970s reporting the photocatalytic activity of ZnO and TiO2 electrodes [1, 2], semiconductor‐based photocatalysis has become a topic at the forefront of technology in various disciplines [3–9], particularly in the fields of solar light conversion for environmental remediation (i.e. wastewater treatment, self‐cleaning surfaces, and air purification purposes) and in the production of energy either via the photovoltaic effect or by the photogeneration of energetic vectors (mainly H2 and O2 from the photochemical water splitting and fuels from the photoreduction of CO2).
Heterogeneous photocatalysis has been mainly associated with the use of wide bandgap n‐type semiconductors as photoactive materials such as titanium oxide [5, 6]. The mechanism of light absorption in these materials is based on the interband electron transitions that occur when photons of adequate energy are adsorbed by the photocatalyst. This photogenerates surface charge carriers (electron/hole pairs) whose fate will determine the efficiency of the photocatalytic reaction (recombination emitting light or dissipating energy as heat, surface migration, and reaction with electron donor or acceptors). For environmental applications in aqueous environments, the efficiency is also often measured as the rate of generation of oxygen‐reactive radical species (mainly HO• and O2−•) that undergo further decomposition reactions, enhancing the efficiency of the overall photocatalytic process.
A major drawback of most semiconductors is that much of sunlight is wasted due to their poor photonic efficiency. As an example, the sunlight exploitation of the benchmark material, TiO2, is lower than c. 4% [10, 11]. However, semiconductors with visible light activity typically suffer from severe photocorrosion phenomena, being instable upon long illumination periods [12, 13]. Other important challenges of most semiconductors toward the large‐scale implementation of heterogeneous photocatalysis in environmental remediation are associated with the high surface recombination rates of the photogenerated electron–hole pairs and the separation (filtration), recovery, and reutilization of the fine semiconductor powders [5, 6, 14, 15].
Numerous efforts have been made in the past decades for overcoming these limitations, most of them based on the development of novel photoactive materials with improved visible‐light‐harvesting features [4, 16, 17], incorporation of dopants to modify the electronic band structure of semiconductors and reduce surface recombination [18–22], surface sensitization with dyes [23, 24], nanosizing the photocatalysts for an efficient charge separation, and the synthesis of porous semiconductors and/or immobilization on inert porous substrates, among most representative [25, 26]. In this regard, extensive work has been carried out to couple semiconductors with a variety of carbon materials of different forms and morphologies and prepared by many different pathways, for enhancing the spectral response of the semiconductor/carbon composites in the UV–visible light range [27–39].
Overall, it is accepted that the role of a carbon material as an additive to a semiconductor greatly differs depending on the structural features of the carbon itself. For instance, for carbons with high electron mobility, such as carbon nanotubes and graphene, the enhanced photocatalytic performance of semiconductor/carbon composites has been mainly attributed to the strong interfacial electronic effects between the carbon material and the semiconductor. This favors the separation of the photogenerated carriers via the delocalization in the π electron density of the carbon matrix, which will be acting as an efficient photosensitizer [28, 32]. In 2009, Luo et al. reported the first evidence on the self‐photoactivity of carbon nanotubes under visible light, attributed to the presence of structural defects and vacancies [40]. Later, due to the unique optical features of graphene, many works have focused on the photoactivity of graphene itself and graphene‐based nanocomposites [32, 34, 38, 39, 41]. However, the beneficial effect of the use of graphene for photocatalytic purposes is still quite controversial and has been recently argued that its role in TiO2/graphene composites on enhancing the photocatalytic activity of TiO2 is in the same essence as that of any other carbon material [39].
Interestingly, despite the structural and chemical similarities between amorphous carbons and graphene, there has been a dearth in exploring the self‐photochemical activity of these forms of carbon. In the particular case of nanoporous carbons, the most studied in the literature are traditionally based on the use of activated carbons of high porous features as inert supports to semiconductors [27, 36, 42, 43]. Indeed, up to 2010, the enhanced photoactivity of semiconductor/activated carbon composites has explained the enhanced mass transfer of the pollutant from the bulk solution due to the porosity of the carbon (the carbon material was considered a non‐photoactive support) [36, 42, 43], coupled with the enhanced photoactivity of the immobilized semiconductor due to the stabilization of the exciton in the graphene sheets of the carbon [27–39].
In 2010, our research group provided the proof of the concept of the photocatalytic activity of semiconductor and metal‐free nanoporous carbons under UV–Vis irradiation [44], as well as the first experimental evidence on their ability to generate reactive radical species when exposed to light in aqueous environments [45, 46]. This study opened up new perspectives on the use of nanoporous carbons as in heterogeneous photocatalysis for the degradation of pollutants, and the topic has become a largely investigated one. Aiming to unravel the origin of the self‐photoactivity of nanoporous carbons, we have investigated the application of nanoporous carbons in several photocatalytic reactions under varied irradiation conditions. These studies have thrown some light on the nature of the carbon/light interactions and their correlation with the performance of the nanoporous carbons as photocatalysts toward different photo‐assisted reactions, as well as the dependence on the properties of the nanoporous carbon (i.e. surface chemistry and average nanopore size) and the characteristics of the irradiation source [44–54].
The aim of this chapter is to provide an overview on the possibilities of exploiting the interactions of nanoporous carbons and different sources of irradiation (UV, simulated solar light) in different fields of research, covering the progress in the photocatalytic degradation of pollutants using metal‐free nanoporous carbons as photocatalysts for environmental remediation (where the photocatalytic activity of the nanoporous carbon adsorbent can be used with a dual purpose of simultaneously promoting the degradation of the pollutant and regenerating the exhausted carbon bed), as well as new perspectives on the use of nanoporous carbons in photochemical applications.
6.2 Semiconductor‐Free Nanoporous Carbons as Photocatalysts
Our studies in 2010 reported the anomalous photochemical activity of a semiconductor‐free nanoporous carbon, showing a significant level of self‐photocatalytic activity under UV–visible irradiation [44–46]. The study showed an improvement in the photooxidation of phenol in aqueous solution using the nanoporous carbon alone as photocatalyst, outperforming the photolytic breakdown and bare or immobilized titania.
Aiming at understanding this behavior and to discriminate whether the observed self‐photochemical activity was a singular characteristic of that particular activated carbon [44, 47] or rather an intrinsic property of carbon materials beyond the so‐called synergistic effect of the porosity widely reported in the literature [28, 29], further photodegradation studies have been extended to other nanoporous carbons.
A screening of various nanoporous carbons obtained from different precursors (i.e. coal, plastic waste, lignocellulosic residues, polymers) showing varied chemical and textural features was considered (Table 6.1) toward the photodegradation of phenol in solution (as model molecule). The performance of a selection of nanoporous carbons under dark conditions and upon irradiation is shown in Figure 6.1, along with data corresponding to direct phenol photolysis for comparison.
Table 6.1 Main physicochemical characteristics of the selected nanoporous carbons obtained from gas adsorption data (i.e. N2 adsorption/desorption isotherms at −196 °C), elemental analysis (on dry ash‐free basis), and pH of the point of zero charge.
| Remarks | SBET (m2 g−1) | VPORES (cm3 g−1) | VMICRO (cm3 g−1) | VMESO (cm3 g−1) | pHPZC | O/N/S (wt%) | References | |
| NC | Coal (steam activation) | 1031 | 0.52 | 0.32 | 0.089 | 8.9 | 1.9/0.7/0.3 | [44, 45, 48] |
| NCG | Sample NC partially graphitized at 2400 °C | 33 | 0.08 | 0.00 | 0.010 | 6.5 | 0.02/—/— | — |
| NC‐deashed | Sample NC after deashing by acid digestion | 1080 | 0.53 | 0.33 | 0.080 | 7.6 | 1.9/0.7/0.3 | [46, 55] |
| NC‐noFe | Sample NC after selective removal of iron traces in the ashes | 1032 | 0.52 | 0.32 | 0.080 | 7.4 | 1.9/0.7/0.3 | [46, 55] |
| NC‐S | Sample NC oxidized in ammonium persulfate 8 h | 989 | 0.50 | 0.31 | 0.07 | 3.3 | 7.5/0.4/0.7 | [51] |
| NC‐SH | Sample NC‐S treated at 400 °C in inert atmosphere | 1045 | 0.51 | 0.32 | 0.08 | 5.7 | 6.4/0.5/0.4 | [51] |
| NC‐S2 | Sample NC oxidized in ammonium persulfate 17 h | 839 | 0.42 | 0.29 | 0.07 | 2.3 | 12.3/—/0.2 | [48] |
| NC‐U | Sample NC‐S2 after urea modification | 945 | 0.53 | 0.35 | 0.08 | 10.5 | 4.5/3.9/4.5 | [48] |
| CV | Biomass (H3PO4 activation) | 1280 | 1.06 | 0.31 | 0.52 | 2.2 | 9.8/—/0.1 | [47, 49] |
| CVH8 | Sample CV after thermal treatment at 850 °C in inert atmosphere | 1040 | 0.79 | 0.27 | 0.36 | 6.5 | 6.0/—/0.1 | [49] |
| BKK | Bituminous coal (steam activation) | 961 | 0.57 | 0.29 | 0.06 | 8.5 | 4.7/0.2/0.8 | [45] |
| A4 | Plastic waste (CO2 activation) | 1357 | 0.60 | 0.45 | 0.04 | 10.9 | 0.6/—/— | [45, 47] |
| LSM | Biomass (steam activation) | 899 | 0.63 | 0.35 | 0.18 | 8.2 | 3.3/—/0.5 | [45, 46] |
| F | Biomass (steam activation) | 799 | 0.35 | 0.26 | 0.03 | 8.1 | 2.9/0.1/— | [45, 47, 50, 52] |
| F‐deashed | Sample F after deashing by acid digestion | 752 | 0.31 | 0.32 | 0.02 | n.a. | 4.0/4.0/— | [56] |
| F1 | Sample F overactivated under CO2 atmosphere for 12 h | 1150 | 0.50 | 0.45 | 0.04 | 8.2 | 3.4/0.1/— | [50, 52] |
| F4 | Sample F overactivated under CO2 atmosphere for 40 h | 1800 | 0.84 | 0.66 | 0.15 | 8.0 | 3.7/0.1/— | [52, 56] |
| F‐S | Sample F modified in H2S | 765 | 0.32 | 0.32 | 0.02 | 7.9 | 3.6/3.6/— | [52, 56] |
| F1‐S | Sample F1 modified in H2S | 1090 | 0.49 | 0.40 | 0.05 | 7.6 | 6.1/5.3/— | [52, 56] |
| F4‐S | Sample F4 modified in H2S | 1535 | 0.75 | 0.55 | 0.13 | 7.4 | 5.5/6.9/— | [52, 56] |
| GEX | Expanded graphite | 8 | 0.02 | 0.002 | 0.001 | n.a. | n.a. | [45] |
| CS | Polysaccharides (hydrothermal carbonization) | 10 | 0.02 | 0.01 | 0.01 | 3.5 | 24.8/—/— | [45] |
| CSH8 | Sample CS treated at 800 °C under N2 atmosphere | 569 | 0.33 | 0.18 | 0.01 | 6.8 | 8.8/—/— | [45] |
| TCSUC | Nanoporous carbon prepared by nanocasting of SBA‐15 using sucrose as precursor | 1394 | 1.25 | 0.31 | 0.81 | n.a. | n.a/—/— | [45] |
| TCCVD | Nanoporous carbon prepared by nanocasting of SBA‐15 by chemical vapor deposition (propylene precursor) | 712 | 0.87 | 0.12 | 0.71 | n.a. | —/—/— | [45] |
n.a. not available.
a Evaluated at p/po 0.99 in the N2 adsorption/desorption isotherms at −196 °C.
b Evaluated from non‐local density functional theory (NLDFT) method by N2 adsorption/desorption isotherms at −196 °C.

Figure 6.1 Concentration decay curves of phenol on various nanoporous carbon photocatalysts under (a) dark and (b) UV irradiation.
Source: Velasco et al. 2012 [47]. Reproduced with permission from Elsevier.
It is important to point out that in the presence of nanoporous catalysts, the photocatalytic degradation becomes a complex phenomenon as several processes may coexist. Indeed, when photons of high energy are used, the concentration of the target pollutant in solution is expected to decrease due to (i) the photolytic breakdown of the compound upon direct interaction with the irradiation (a reaction dependent on the pollutant concentration in solution) and (ii) the adsorption on the porosity of the catalyst (the adsorbed fraction increases with time, thereby modifying the solution concentration of the pollutant), a non‐degradative process that strongly depends on both the porous features of the carbon and the affinity of the pollutant toward the carbon adsorbent. Finally, it should also be considered that the kinetics and yield of the photolytic reaction depend on the solution concentration of the pollutant, which is varying on the course of the photocatalytic tests based on the adsorption kinetics. As a result, the solution gradually turns into a complex multicomponent matrix due to the appearance of the degradation derivatives. These may undergo further photodegradation reactions and/or be adsorbed in the porosity of the carbons, making the scenario a complex one. Hence, to differentiate between the adsorptive and the photooxidative processes (where both adsorption and degradation occur simultaneously), photocatalytic experiments using the nanoporous carbons as catalysts need to be compared to those under dark conditions.
As seen in Figure 6.1, the photolytic breakdown of phenol can be quite high, c. 78% for a solution of 100 ppm of phenol irradiated for 6 h using a 125 W Hg lamp [47]. However, the degradation efficiency in terms of total carbon content under these experimental conditions is below 3%. This is due to the fact that phenol intermediates are not further photooxidized in the absence of a catalyst. However, the removal of phenol in dark conditions is due to adsorption on the carbons' porosity (degradation in the dark was not observed in the timescale of the catalytic tests, 6 h). As expected, the amount adsorbed at dark conditions was controlled by the physicochemical characteristics of the carbons, with the largest uptakes for the carbons with higher surface area and micropore volumes and hydrophobic character (Table 6.1). On the contrary, a low uptake was obtained for carbons of acidic nature, regardless of the porous features [47].
For most of the studied carbons, the overall rate of phenol disappearance remarkably increased under UV–Vis irradiation compared to dark conditions, confirming the photoactivity of the series of nanoporous carbons in the absence of a semiconductor. This trend was more evident at short illumination times, particularly for the nanoporous carbons of hydrophilic character (sample CV), where phenol uptake was not favored [47, 57, 58]. Another important observation was that the rate of phenol disappearance under UV light (encompassing both photooxidation and adsorption) was connected to the porous features of the nanoporous carbons, with slow kinetics for carbons with a narrow distribution of micropore sizes.
Interestingly, the study revealed that some nanoporous carbons (sample A4) exhibited similar performance for phenol degradation in dark and under illumination [47]. At converse, this particular nanoporous carbon showed a negative response of the photogeneration of radicals under illumination [45] but presented certain extent of photochemical activity for other different reactions (i.e. photoelectrochemical water splitting) [56]. This suggested that the photocatalytic activity of nanoporous carbons is not an inherent property of these materials, but it is rather related to their physicochemical and/or structural features, as well on the studied reaction. More interestingly, this observation might explain why all previous studies in the literature had reported the non‐photoactive behavior of activated carbons [29, 36, 42, 43].
More importantly, the evolution of the intermediates concentration in solution with irradiation time was also strongly dependent on the characteristics of the nanoporous carbons. In the absence of a photocatalysts, phenol conversion is quite high and proceeds through the formation of dihydroxylated compounds (catechol, hydroquinone, and benzoquinone), the oxidation in ortho and para positions being the main intermediates. The relative abundance of the degradation subproducts detected in solution changed significantly when the photocatalytic reaction was carried out in the presence of the nanoporous carbons, with a marked regioselectivity for the formation of catechol over quinones (as opposed to titania benchmark). This degradation pathway is considered more advantageous for the overall reaction yield as it proceeds through a mechanism involving a lower number of subproducts (organic acids) [59].
All these results suggested several possible scenarios to explain the differences in overall conversion (rate and intermediates speciation) under dark and UV irradiation, on which the increased degradation rates and yields are linked to [47]: (1) the effect of the solution concentration on photolysis, (2) the adsorption/desorption of the pollutant and photooxidation intermediates on the porosity of the carbon, enhancing the conversion due to the change in the pollutant concentration over time, (3) the occurrence of carbon/light interactions that would provoke the degradation of the adsorbed compounds retained in the pores of the carbons, and (4) the formation of radical species upon illumination of the carbons in an aqueous medium.
The contribution of scenario (1) could not be completely discarded due to the complexity of the solution as a result of the large contribution of phenol photolytic degradation. However, two facts should be considered: (i) under these experimental conditions, none of the studied nanoporous carbons reached their adsorption saturation capacity during the photocatalytic runs, and (ii) the adsorption behavior of phenol and its photooxidation intermediates on nanoporous carbons are similar. In other words, the nanoporous carbons have enough adsorption sites to account for the complete removal of phenol and photooxidation subproducts, despite of which they are detected in solution during the photocatalytic runs.
Furthermore, the analysis of the porous features and thermogravimetric profiles of the carbon samples after the photocatalytic tests revealed interesting information to rule out the contributions of scenarios (1) and (2). Indeed, thermogravimetric curves of the nanoporous carbons after the photocatalytic tests rendered similar profiles than those corresponding to the dark experiments. Analogously, the porous features of the carbons after the photocatalytic runs displayed slightly higher porous features than those of the carbons after adsorption at dark conditions. Based on the higher phenol disappearance rates under UV light, one would have expected just the opposite trend if all phenol were only retained on the porosity of the carbons. The mass unbalance between the amount of compounds detected in solution and that retained on the carbon porosity after the photocatalytic runs cannot be explained by the confinement of the pollutant in the porosity (scenario 2) but rather to the degradation of phenol, thereby confirming the photocatalytic activity of the studied carbons for this particular reaction.
Besides phenol, the intrinsic photocatalytic activity of nanoporous carbons has also been investigated toward the degradation of other compounds such as pharmaceuticals or water (to produce hydrogen and oxygen). For instance, Velo‐Gala et al. reported the photooxidation of sodium diatrizoate (a recalcitrant pollutant widely used in hospitals as iodinated contrast and detected in urban wastewaters, surface waters, and ground waters) under UV light using several commercial and modified activated carbons [60, 61]. For all the studied carbons, the photodegradation rate of sodium diatrizoate (excluding direct photolysis and adsorption) was enhanced in the presence of the nanoporous carbons. Such improvement was found to be dependent on the chemical functionalization of the activated carbon [60, 61]. Additionally, metal‐free nanoporous carbons have also been explored as photoanodes in the photoelectrochemical water splitting, with quite remarkable photocurrent values at low overpotentials under solar irradiation [53, 62, 63].
6.3 Pollutant Confinement on the Porosity of the Nanoporous Carbons
To demonstrate the occurrence of light/carbon interactions eventually leading to the photooxidation of the pollutants retained inside the porosity of the carbons (scenario 3) and discriminate the contributions of other secondary reactions, a new approach was developed to monitor the photocatalytic reaction from a different viewpoint: inside the nanopores of the carbon photocatalysts. This consisted on carrying out the irradiation of the nanoporous carbons previously loaded with the target pollutant and monitoring the photodegradation reaction both inside the nanopores and in the aqueous phase (Figure 6.2a). This novel approach allowed neglecting the effects of adsorption kinetics and solution photolysis and unambiguously demonstrates that semiconductor‐free nanoporous carbons alone are capable of a significant level of self‐photoactivity under adequate irradiation [47, 48, 55].

Figure 6.2 (a) Schematic representation of the experimental approaches designed for evaluating the photocatalytic activity inside the nanoporosity of carbon materials; (b) Quantification of the species detected in the extracts of a series of nanoporous carbons preloaded with 42 mmol phenol after UV irradiation for 20 min; (c) Correlation of phenol conversion with selected physicochemical properties of the nanoporous carbons.
Briefly, the nanoporous carbons were loaded by putting in contact an adequate amount of the carbon with phenol solution until complete adsorption. Subsequently, the suspensions of the loaded carbon with water were irradiated. The extent of the reaction was evaluated from the mass balance of phenol and intermediates detected from both the aqueous solution and inside the nanoporous carbon (by solvent extraction). To avoid the eventual desorption of phenol and/or intermediates during the photocatalytic tests, the concentration of phenol loaded in the porosity of the carbon was adjusted well below the saturation limit of the nanoporous carbons. Indeed, no lixiviation of either phenol or its degradation subproducts toward the solution was detected at any time for any of the studied carbons. Thus, the extent of phenol conversion corresponds exclusively to the degradation inside the porosity of the carbon.
Figure 6.2b shows the data obtained for the photooxidation of phenol determined upon irradiation of aqueous suspensions of the preloaded nanoporous carbons. As seen, for most of the investigated carbons, a significant fraction of the adsorbed phenol molecules was decomposed upon light exposure, being the overall phenol conversion larger or similar than in the photolytic reaction (benchmark used for comparison purposes). This is most remarkable considering the strong light shielding effect of the carbon matrix, which is expected to reduce the photon flux arriving at the phenol molecules adsorbed inside the pores. Interestingly, one of the carbons showed a negative response with a negligible photodegradation of phenol inside the porosity. Regarding the distribution and nature of the intermediates, outstanding differences were seen among the nanoporous carbons. All this suggests that the photochemical response of nanoporous carbons does not only depend on the confinement of the pollutant but also seems to be related to the nature of the carbon, most likely composition, surface acidity/basicity, structure, etc. However, no straightforward correlation has been found between the photochemical response of the nanoporous carbons toward phenol degradation and selected physicochemical properties such as the specific surface area, pore volume, composition, or surface acidity (Figure 6.2c). Nevertheless, data corroborated that when selected nanoporous carbons are illuminated, carbon/photons interactions take place, leading to the formation and propagation of charge carriers that seem to have enough redox potential to directly oxidize phenol (scenario 3) and/or generate reactive species (scenario 4).
In an attempt to quantify the efficiency of phenol photodegradation inside the nanopore space of the carbons, the photochemical quantum yield of phenol (Φ) – defined by IUPAC as the ratio between the number of moles reacted, ΔN, per mole of photons absorbed (IAΔt) – was determined. This parameter is evaluated from the slope of the relation between the moles of pollutant degraded per incident photon flux versus the irradiation time with the equation [64]:
where IA is the photon flux absorbed by the sample, evaluated from the product of the incident photon flux Io provided by the lamp (determined by actinometry), and the integrated absorption fraction over the wavelength range used in the experiment.

Figure 6.3 (a) Phenol degraded per mole of absorbed photons with the irradiation time in a series of preloaded nanoporous carbons with varied chemical composition. (b) Pseudo photochemical quantum yields (ΦPS) for two different regimes.
Considering the contributions of light scattering by the catalyst particles suspended in solution and the fraction of light absorbed by the carbon matrix, the evaluation of the accurate incident photon flux reaching the molecules adsorbed inside the pores (needed for the evaluation of the quantum yield) is very complex. Thus, as an approximation, a pseudo photochemical quantum yield (ΦPS) was proposed, evaluated considering that the photon flux provided by the lamp can reach the molecules adsorbed in the pores of the carbon catalyst, and, therefore, can be absorbed and used in the photo‐assisted reaction (i.e. IA ∼ Io). This approximation cannot be considered sensu stricto a quantum yield, because the actual photon flux reaching the adsorbed molecules in the pores is certainly much lower than the incident flux provided by the lamp (due to the above‐mentioned contributions of light scattering and carbon light absorption); however, our approximation would account for the minimum limit of the actual quantum yield, allowing the comparison of the different carbons.
The pseudo‐quantum yields retrieved for a series of nanoporous carbons with varied composition (Table 6.1) are shown in Figure 6.3, compared to data from photolysis in solution. In the absence of a photocatalyst, the real incident photon flux can be easily measured by chemical actinometry, allowing an accurate determination of the photochemical quantum yield (Φ) for phenol degradation. As seen, the plot of the moles of degraded phenol per incident photon flux versus the irradiation time is a straight line, yielding Φ = 11 mmol/einstein, in good agreement with the values reported in the literature for phenol photolytic degradation [65, 66]. The ΦPS values estimated for the nanoporous carbons showed two different regimes; below 30 min of irradiation, the plots were very steep with ΦPS close to unity; at longer illumination times, a second linear region with a smoother slope was obtained, with lower values of ΦPS compared to those of the first regime (although still about five times higher than the value obtained for the solution photolysis). These two regimes suggest a fast consumption of the photogenerated charge carriers in the first stage (likely related with consumption of water and/or oxygen entrapped in the pores), leading to a diffusion‐limited rate in the second stage of the photodegradation reaction or a partial deactivation or consumption of some photoactive sites of the nanoporous carbon [47, 67].

Figure 6.4 Phenol conversion normalized per incident photon flux for various wavelengths on a series of nanoporous carbons.
Source: Gomis‐Berenguer et al. 2017 [54]. Reproduced with permission from Elsevier.
6.3.1 Effect of Pore Size and Wavelength Dependence
To further comprehend the effect of nanoconfinement and its dependence with the nature of the characteristics of the nanoporous carbons and the wavelength of the irradiation source, a series of nanoporous carbons exhibiting controlled porous features [52, 54] were investigated using monochromatic light. Unlike polychromatic light, this allows to evaluate the impact of energy of the incident photons on the photochemical conversion without biased interpretations.
Figure 6.4 shows the wavelength dependence of the photodegradation of phenol inside the porosity of various carbon materials. The photoconversion values appear normalized per incident photon flux to allow a direct comparison of all data for each wavelength.
In all cases, phenol conversion inside the carbon nanopores was higher than that of photolysis, corroborating the outstanding role of nanoconfinement in enhancing the light conversion into a chemical reaction. With one exception, data confirmed the photochemical activity of the studied nanoporous carbons at various wavelengths between 200 and 600 nm. The performance followed a U‐shaped pattern with the wavelength, with a minimum of conversion between 400 and 450 nm. A similar trend was obtained for other series of nanoporous carbons and using different lamp set‐ups [48], to discard any effects from eventual low‐incident photon flux. Most importantly, the onset of phenol photooxidation reaction was measured at 432 nm for the nanoporous carbons, which corresponds to over 100 nm redshift compared to the non‐catalyzed reaction. Additionally, a significant photochemical activity at wavelengths above 400 nm – corresponding to the visible range – was observed for most of the studied carbons.
To gain additional insight on the dependence of the yield of the photochemical reaction with the nanopore size and the confinement state of the target molecule, a second series of nanoporous carbons showing a gradual modification of the average micropore size were studied. The starting carbon (sample F) was a biomass‐derived material displaying a narrow distribution of pore sizes within the micropore range as evidenced from gas adsorption data (Table 6.1). To explore the dependence of the photoactivity with the average pore size, sample F was activated under CO2 atmosphere for variable periods of time to obtain samples F1 and F4 with increasing burn‐off degrees. Owing to the strictly controlled activation conditions, the chemical composition of the pristine carbon remained almost unaltered (as further confirmed by additional characterization of the samples by X‐ray photoelectron spectroscopy (XPS) and temperature‐programmed desorption‐mass spectrometry (TPD‐MS)), and the changes exclusively affected the porous features of the carbons (i.e. pore volume, surface area, and particularly the narrow microporosity) [52]. More precisely, the activation brought about a gradual enlargement of the micropores, which are known to be the active sites for phenol adsorption on nanoporous carbons. This is important as the photocatalytic reaction was carried out in the preloaded carbons.
Interesting features were revealed from the analysis of phenol conversion values at selected wavelengths of the series of carbons with increasing burn‐off degrees (Figure 6.5); the experimental non‐normalized values at each wavelength are also presented for clarity. As also reported for other carbons, the photochemical performance followed a U‐shaped pattern with the wavelength, with a minimum featuring at 400 nm – derived from very low conversion values – regardless of the activation conditions. At wavelengths corresponding to visible light (i.e. 450 and 500 nm), the conversion rose again with values very close to those obtained at 269 nm.
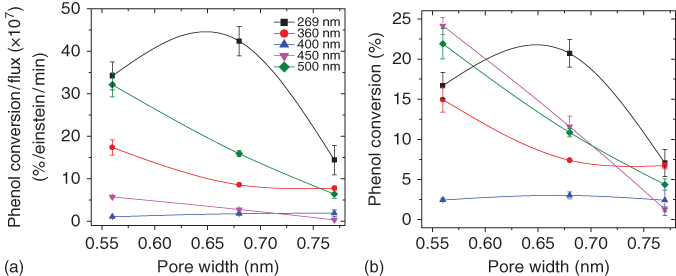
Figure 6.5 Dependence of phenol photooxidation conversions with the average micropore size evaluated from gas adsorption data; (a) normalized conversion versus incident photon flux; (b) absolute conversion.
Source: Gomis‐Berenguer et al. 2016 [52]. Reproduced with permission from Elsevier.
However, a clear correlation was obtained between the conversion and the confinement state of adsorbed phenol molecules. As a general trend, the photochemical conversion of phenol gradually decreased with the enlargement of the average micropore size, being the effect less remarkable at high wavelengths. The degradation of the intermediates was more efficient at 269 nm, pointing out the difficulty in pursuing the oxidation of dihydroxylated aromatic compounds at high wavelengths corresponding to the visible light range. Data also showed that there is a threshold pore size for an optimum exploitation of the light, which is connected with the dimensions of the target molecule retained in the porosity and the strength of the interactions between the pollutant and the nanopore walls. At 269 nm, the light conversion for phenol oxidation was enhanced for pores below 0.7 nm, whereas the photochemical response was drastically reduced when the pores were widened by 0.1 nm. Similar patterns were obtained for higher wavelengths, although the drop in the conversion was pronounced (Figure 6.5). This trend was attributed to the tight confinement of phenol molecules in the narrow microporosity of the carbon, maximizing host–guest interactions. When the pore aperture was enlarged, the interactions between the adsorbed phenol molecules and the pore walls became weaker [57, 58], thus decreasing the probability of the fast charge transfer of the photogenerated carriers. In this situation, other electron donors and/or hole scavengers present in the medium (i.e. oxygen and water molecules) compete with adsorbed phenol molecules, resulting in lower conversions.
6.3.2 Effect of Functionalization with O‐, N‐, and S‐Containing Groups
Another important factor that controls the photocatalytic performance of nanoporous carbons is the chemical composition, and most importantly, the type and density of surface groups present on the carbons. To explore the effect of surface functionalization of nanoporous carbons on their photocatalytic performance, a series of carbons were chemically modified under mild conditions to introduce O‐, N‐, and S‐containing groups [48, 50, 51]. As the chemical modification did not modify the textural parameters of the carbons, the changes in the photocatalytic reaction inside the nanoconfined pore voids of the carbons were exclusively associated with the surface functionalization (Table 6.1).
The incorporation of N and O groups (via wet oxidation with nitric acid and N modification by thermal treatment in urea) slightly modified the photoconversion of phenol using polychromatic light, following the trend: pristine ∼ N‐doped > O‐doped carbon (samples NC, NC‐U, and NC‐S2, respectively). Oxidation of carbon decreased the conversion of both phenol and degradation intermediates [48], indicating that the overall photooxidation process is more efficient on carbons of hydrophobic nature. This contrasts with other studies in the literature, reporting an increased performance toward oxidation of other pollutants after the incorporation of O groups in the carbon matrix [33]. This apparent contradictory result was explained by the impact of oxidation treatment on the interactions between carbon material and target pollutant, indicating that these interactions govern the conversion inside the porosity (c. oxidation of carbon results in lower phenol affinity).
The most interesting finding of this study was the wavelength dependence of the photochemical response after the chemical modification. Although the oxidized carbon showed similar wavelength dependence than the pristine material, N doping (mainly in the form of quaternary nitrogen and pyridine‐type groups) provoked a redshift in the light absorption features (seen in the increased conversion at 432 and 545 nm). These differences were explained in terms of changes in the π‐electron density of graphene sheets of the carbon matrix upon functionalization.
In the case of N functionalization, nitrogen moieties are known to increase the polarity and the electron transfer properties of carbon due to electron‐donating character of the lone‐pair of electrons of the nitrogen atoms [68]. This is expected to modify the density of electronic states (DOS) of the carbon material, likely by introducing intermediate states of higher energy between the conduction and the valence band [69, 70]. Such intermediate states would act as stepping stones facilitating the absorption of low‐energy photons, accounting for the observed photochemical activity at wavelengths corresponding to visible light. For O groups, the fall in phenol photooxidative conversion can be attributed to the electron withdrawal effect of the functionalization on carbon, which would affect the stabilization of photogenerated carriers by delocalization within the carbon basal planes.
To gain further insight on the effect of O functionalization, the phenol conversion percentages of a series of nanoporous carbons with various oxidation degrees were evaluated. Figure 6.6 shows the dependence of phenol conversion with selected parameters indicating the degree of surface functionalization of the studied carbons (as oxygen content, surface pH, and the nature of the O surface groups). A close look of the data clearly indicates that the incorporation of oxygen groups of acidic nature is the most determinant parameter in the reduction of photochemical activity of the nanoporous carbons. This was also supported by the correlations with the amounts of gases (CO and CO2) quantified from the thermodesorbed species by TPD‐MS [51, 54]. For instance, the higher performance of carbon NC‐SH (slightly oxidized) compared to NC‐S (heavily oxidized) is due to its increased surface hydrophobicity (Table 6.1). For both carbons, the amount of CO‐releasing groups (mainly phenolic and quinone‐type groups) is quite similar, whereas the amount of CO2‐evolving groups (attributed to carboxylic acids and anhydrides of acidic nature) is about three times higher in NC‐S than in NC‐SH. A similar trend was observed when the global number of moles reacted was considered, rather than just phenol conversion. In this case, the dependence followed a polynomial pattern, indicating a more sensitive response of the light conversion yield to small changes in the surface acidity of the nanoporous carbons.

Figure 6.6 Dependence of phenol conversion of samples NC, NC‐S, and NC‐SH with (a) oxygen content and pH of the point of zero charge and (b) the amounts of CO and CO2 released from carbons as detected by TPD‐MS.
Source: Velasco et al. 2015 [51]. Reproduced with permission from John Wiley & Sons.
For the incorporation of S groups (Figure 6.7), the general trend showed an improved photoactivity with the sulfur content at all the wavelengths, particularly at 269 and 500 nm, demonstrating the enhanced exploitation of light in the UV and visible ranges [50]. The photochemical response of the carbons was strongly influenced by the chemical environment of the S groups, with oxidized forms of sulfur being more photoactive than thiols and/or sulfides (Figure 6.7b). A positive effect of S functionalization has also been reported for a different reaction: photoelectrochemical splitting of water using polychromatic light [62, 63].

Figure 6.7 (a) Effect of sulfur on the conversion of phenol of the series of carbons with gradually increasing micropore sizes defined as the difference between the conversion in the S‐doped and its corresponding as‐received carbon. (b) Relative abundance of S moieties on the nanoporous carbons detected by deconvolution of TPD‐MS profiles (m/z 48).
Source: Gomis‐Berenguer et al. 2016 [50]. Reproduced with permission from Elsevier.
Several scenarios have been discussed to account for the improved photochemical activity of the S‐functionalized carbons. First, incorporation of sulfur was found to provoke narrowing of the average pore size, thereby boosting the separation of the photogenerated charge carriers by increasing the probability of the reaction with the adsorbed phenol molecules (trapped in the pores) [50]. The fast charge transfer is favored in S‐functionalized carbons, as the presence of heteroatoms is known to lower the energy difference between the electronic levels of the carbons [68]. Second, the stabilization of the holes through the oxidation of water adsorbed in the pores is also expected as was corroborated by electron spin resonance (ESR) spectroscopic studies [50]. Additionally, based on the role of the N‐, S‐, and O‐containing groups, we also proposed some other transitions involving the activation of chromophoric groups on the carbon surface under visible light [45, 46, 48, 50] (see discussion below about the mechanism).
6.3.3 Effect of Mineral Matter
Very often, nanoporous carbons contain small amounts of inorganic impurities (ashes), associated with the carbon precursor or the synthetic method used in the manufacturing process. The ashes are typically considered as inert matter; however, the presence of traces of inorganic elements might play a key role in catalytic and photocatalytic applications, where such impurities might also be active (photo)catalytic sites for a given reaction.
To investigate the effect of the ash content of nanoporous carbons in their photochemical activity, a demineralization treatment was carried out on selected carbons (samples NC and F), showing a higher photocatalytic activity toward the photooxidation of phenol. An acid digestion using HF and HCl acids at moderate temperature [71] allowed the complete removal of the inorganic impurities of both carbons, without any other modification of the texture (as inferred from the gas adsorption data) or the chemical composition beyond the elimination of the ashes (i.e. no oxidation of the carbonaceous matrix, as inferred from elemental analysis). The deashed nanoporous carbons (F‐deashed and NC‐deashed samples in Table 6.1) were used as photocatalysts toward phenol photooxidation using monochromatic (Figure 6.8) and polychromatic light. In the case of polychromatic light, data showed a decrease in the performance of the deashed carbons, with values of phenol conversion ranging from 92% to 83% for carbons F and F‐deashed and from 98% to 93% for NC and NC‐deashed, respectively [55, 56]. Still the conversion values of the deashed carbons were higher than those corresponding to the photolytic degradation of phenol in solution (c. 80%). This unambiguously demonstrated that even if the inorganic matrix of the carbons may contribute to some extent to the observed photoactivity toward the oxidation of phenol in solution, the carbon matrix itself also presents a non‐negligible photoactivity.

Figure 6.8 Phenol conversion per incident flux as a function of the wavelength of the irradiation source for the pristine and deashed carbons. Photolysis values are included for comparison purposes.
The fact that the inorganic matters do not contribute to decrease the photochemical performance of the nanoporous carbons constitutes an important finding from a technological viewpoint, as it would allow the direct implementation of this materials in photocatalytic applications at a large scale without the need to remove the ashes.
As for the wavelength dependence, different behaviors were obtained for the studied nanoporous carbons. In the case of carbon NC, the general trend showed that the conversion of phenol gradually decreased with rising the wavelength of the irradiation source, reaching very low values for 400 nm (absolute conversion values before photon flux normalization ranging from 20% at 254 nm to 1% at 400 nm) and no conversion at 450 nm. The low absolute conversions measured above 400 nm suggested the lack of light/carbon matrix interactions upon irradiation at these conditions (photon fluxes at these wavelengths were not among the lowest recorded values for the investigated wavelengths).
In the case of carbon F, an irradiation source with a distinct distribution of intensity was used to further clarify the performance of the deashed carbons under visible light. Regarding the effect of the mineral matter, a similar trend was observed compared to carbon NC, with a slightly lower photoactivity of the carbon after the removal of the ashes. However, the wavelength dependence was markedly different for both nanoporous carbons. Indeed, for carbons F and F‐deashed irradiated at 269 and 500 nm, the normalized phenol conversion was reduced by c. 20% and 14%, respectively. Notwithstanding, for 360, 400, and 450 nm, the diminution of degradation was less remarkable. Despite the fall, deashed carbon still showed quite a large phenol photoconversion, particularly at high and low wavelengths. This indicates that, although there is a non‐negligible photocatalytic activity of inorganic matter, ash‐free carbon is able to promote phenol photooxidation inside the porosity of the carbon when exposed to irradiation.
6.4 Postulated Mechanisms
Based on the experimental evidence on the photochemical activity of nanoporous carbons of different physicochemical features (texture, composition, and structure), gathered under varied illumination conditions for the photooxidation of pollutants in aqueous environment, several hypotheses have been proposed to account for the origin and nature of the carbon/light interactions and the exploitation of light conversion in nanoconfined carbon pore spaces.
The absorption features of amorphous carbons have been reported to depend on the DOS, whose energy spread of states depends on the sp2:sp3 hybridization ratio of the carbon atoms, and the electronic transitions involving the sp2 carbon clusters [69, 70, 72–75] and/or the activation of chromophoric groups on the carbon surface [62, 63]. The size and ordering of the sp2 clusters, their population in the carbon matrix, and the creation of distortions of π* states have been suggested to have a strong effect in the optical characteristics of the carbons. Upon irradiation of carbons, these electronic transitions generate excitons (holes or electrons) that can be delocalized through the sp2‐conjugated domains of basal planes of the nanoporous, avoiding or delaying recombination phenomena. This favors their participation in some other electronic transitions with acceptors/donors present in the medium (such as water, oxygen, surface groups, and pollutants), either involving direct hole/electron scavenging or radical mediated mechanisms. A schematic representation of these electronic transitions is illustrated in Figure 6.9.

Figure 6.9 Schematic representation of the mechanism proposed for the exciton formation and fate upon irradiation of nanoporous carbons.
Source: Gomis‐Berenguer et al. 2017 [54]. Reproduced with permission from Elsevier.
Regarding the nature of the photogenerated charge carriers, in the UV range, medium‐to‐low‐range excitons (Frenkel‐like created in the π–π* and σ–π electronic transitions involving zigzag, carbine‐like sites and other intermediate states) [69, 70, 72–75] are expected to be dominant in nanoporous carbons of low functionalization [54, 76], which typically display low dielectric constants compared to n‐type semiconductors as TiO2 [77]. This is consistent with the behavior reported for other carbon nanostructures such as carbon blacks, graphene, or fullerenes of higher polarity and electron mobility, based on the experimental observations and molecular simulations [75, 78, 79].
In functionalized carbons, electron transfer is facilitated as the energy difference between the electronic levels is lower than that in pure carbon units [68], and charge transfer excitons formed by localized states involving O, S, and C atoms may also be formed. Additionally, N‐, S‐, and O‐containing groups can act as chromophores that may be activated under sunlight irradiation, photogenerating vacancies (holes) able to accept electrons from oxygen in water molecules excitons that may participate in further photochemical reactions as proposed for various oxidized forms of sulfur (thioesters and sulfones) and O‐containing groups [79, 80]. The nature of the surface moieties of the nanoporous carbons is also important; for instance, bulky oxidized sulfur moieties (i.e. thioesters and sulfones) located at the entrance of the wide micropores seem to be more photoactive under visible light than thiols and/or sulfides groups [50, 56].
Hence, the varied chemical environments of the surface functionalities present on nanoporous carbons, as well as their different distribution of pore sizes, can explain differences in their photochemical response. On the one hand, light conversion in nanoporous carbons is boosted in the tight nanopore space, pointing out the outstanding role of the confinement state of the adsorbed molecules. Weak molecule/pore wall interactions in large nanopores decrease the probability of splitting of the photogenerated exciton through fast charge transfer reactions with electron donors and/or hole scavengers (i.e. oxygen, adsorbed phenol, and water molecules to form radicals) [45, 46].
On the other hand, the fall in the photochemical conversion in functionalized carbons has been associated with several factors such as the creation of structural defects, changes in the sp2/sp3 hybridization state of carbon atoms during the functionalization treatment, or the lower stabilization of the photogenerated carriers due to the withdrawal effect of surface groups (such as carboxylic acid and anhydrides) on the p‐electron density of the conjugated sp2 network.
The hypothesis of stabilization of the holes through oxidation of water adsorbed in the nanopores of carbons to form reactive oxygen species (ROS) (scenario 4) was validated by ESR spectroscopy using nitrones as chemical trapping agents (e.g. 5,5‐dimethylpyrroline‐N‐oxide (DMPO) and 5‐diethoxyphosphoryl‐5‐methyl‐1‐pyrroline‐N‐oxide (DEPMPO)) [45, 46, 55]. A variety of carbon materials obtained from various precursors and varied chemical, structural, and textural features (i.e. amorphous nanoporous carbons, carbon nanotubes, expanded graphite, carbon hydrochars, and ordered nanoporous carbons) were investigated by irradiation of carbon suspensions in water (Figure 6.10).

Figure 6.10 (a) Comparative of the ESR signals obtained upon 20 min irradiation of aqueous suspensions of selected carbon materials; (b–d) Quantification of the radical species detected from the ESR signals corresponding to DMPO‐OH adducts (second peak intensity in the 1 : 2 : 2 : 1 patterns, marked as *).
Most of the studied carbons showed similar ESR patterns, with the characteristic quartet peak profile with a 1 : 2 : 2 : 1 pattern of intensities corresponding to DMPO‐OH adduct [81]. Some other patterns associated with HDMPO‐OH (2‐hydroxy‐5,5‐dimethyl‐1‐pyrroline‐N‐oxide, HDMPO), DMPO‐R (carbon‐centered radical), and DMPO‐OOH adducts (marked as • in Figure 6.10a) were also identified for some carbons [45, 46]. Detection of DMPO adducts confirmed the formation of hydroxyl and/or superoxide radicals upon irradiation of nanoporous carbons in aqueous medium (scenario 4), upon stabilization of the photogenerated charge carriers. The stabilization of holes through oxidation of water would form hydroxyl radicals, while the electrons participate in the multi‐electron reduction of molecular oxygen to produce superoxide radicals and hydrogen peroxide, which may be further reduced to hydroxyl radical (HO•) or water. Although the ability to photogenerate radicals is well established for conventional semiconductors [82], this issue was not clear for nanoporous carbons. Furthermore, similar findings in the formation of hydroxyl and superoxide radicals upon irradiation of nanoporous carbons have recently been reported by other authors using different chemical trapping agents, various carbon loadings, and reaction times [83].
Quantification of abundance of radical species showed higher DMPO‐OH concentration levels for certain carbons than those corresponding to TiO2 as a benchmark semiconductor under similar experimental conditions (Figure 6.10b–d). Additionally, lower concentration levels of radicals were obtained for the functionalized carbons (Figure 6.10), with a clear correlation between the formation of radical species and the acidic/basic nature of nanoporous carbons. As an example, the amount of ROS correlates well with the presence of CO‐evolving groups of basic nature, while strong acidic groups (CO2 evolving) seem to inhibit the formation of radicals. However, for some carbons, no radicals were detected by ESR (sample A4), which seemed to be in agreement with the lack of photoactivity observed for this material in previous works [47], and demonstrated that formation of photoinduced radicals is not an intrinsic property of all carbon materials.
It should be mentioned that even though ESR spectroscopy may contribute to the understanding of the photochemical response of carbon materials, it just provides a diagnostic indication of formation of free radicals. In this regard, some functionalized carbons showing low concentration of radical species (sample CV) displayed among the highest photocatalytic activity toward phenol photooxidation [45, 55]. This phenomenon has also been described for some titanium oxide powders [84]. Hence, low ESR signals should not be considered as an indication of low photoactivity, but rather to the predominance of a radical‐ mediated or direct hole transfer degradation mechanisms.
6.5 Photocatalytic Cycles
With the purpose of evaluating if nanoporous carbons as photocatalysts fulfill the requirements of long life cycle and good degradation efficiency, the performance of selected nanoporous carbons during consecutive phenol photodegradation runs was investigated [49]. For the study, irradiation was carried out on the suspensions of the carbons preloaded with phenol, maintaining the same initial concentration of phenol in solution for each cycle, to avoid differences in the photolytic reaction.
The photocatalytic performance of the nanoporous carbons gradually decreased with the number of the cycles, the effect being more notorious in the carbon of hydrophilic nature. After the third run, the overall phenol efficiency reached a plateau at about 50% conversion after the first 90 min of irradiation in every run. This is in agreement with the accumulation of intermediates observed for the first cycles, where it seems that the materials are capable of promoting the photodegradation of phenol but not that of the polyhydroxylated intermediates.
Data also showed a marked accumulation of phenol degradation subproducts in the solution during the consecutive photocatalytic runs. The lower photooxidation extent of polyhydroxylated intermediates – compared to phenol itself – indicates the preferential reactivity of the radicals toward the monosubstituted aromatic ring [85], which is a characteristic of electrophilic mediated mechanisms. The higher concentrations of catechol over the cycles confirmed the regioselective oxidation mechanism via the preferential substitution in ortho‐ (i.e. catechol) over meta‐ or para‐ (i.e. quinones) position. Furthermore, the cycleability of the nanoporous carbon photocatalysts was found to depend on their basic/acidic nature, with a somewhat lower performance for the hydrophilic carbons. When similar cycleability tests were carried out using commercial titanium oxide as photocatalyst, lower amounts of aromatic intermediates were detected in solution. However, the accumulation of short alkyl chain organic acids also become important, also leading to poor mineralization values upon consecutive cycles.
Conversion yields and mineralization rates were greatly enhanced in the presence of excess of dissolved oxygen in the solution, demonstrating the outstanding role of oxygen in the photooxidation of phenol and intermediates. This was a critical factor in the long‐term performance of the hydrophilic nanoporous carbon, which showed a sharp fall in conversion upon cycling under oxygen depletion conditions. The evolution of the porous features of the nanoporous carbons upon consecutive cycles also depended on the presence of dissolved oxygen, with a lower collapse of the porosity over cycles under oxygen excess (indicating a higher photocatalytic performance).
6.6 Summary and Conclusions
The discovery of the photochemical activity of metal‐free nanoporous carbons in aqueous environments, due to their ability to photogenerate oxygen‐based radical species when exposed to UV or sunlight, offers new perspectives in the field of applied photochemistry in nanoconfined systems (beyond their use as supports of semiconductors).
The dependence of the photochemical activity of nanoporous carbons with the pore confinement and surface chemistry herein described toward the photooxidation of phenol has set the basis for the implementation of advanced oxidation processes based on carbon photocatalysts for water treatment. This is also expected to be valuable, in general, for targeting other applications in the field of solar energy conversion (e.g. water splitting, carbon photofixation reactions, and photovoltaic devices).
The dual nature of nanoporous carbons as photocatalysts with well‐defined pore architectures and composition is essential for an enhanced light conversion in nanoconfined spaces. The tight nanoconfinement inside the pores is important as the proximity between the photogenerated charge carriers and the adsorbed molecule in the constrained nanopore space facilitates the exciton splitting and charge separation, minimizing surface recombination. Furthermore, matching of the molecular dimensions of the target compound confined in the carbon porous network is an interesting approach to increase the photochemical response of the carbon, while boosting the light conversion toward the visible spectrum (c. about 100 nm redshift). Surface functionalization of the nanoporous carbon is also important, as it may affect the lifetime of the photogenerated species, the light absorbing feature of the carbon material, and the electron acceptor capacity of the carbon matrix. Additionally, some surface moieties might also act as chromophores absorbing light and photogenerating additional charge carriers, thereby enhancing the photochemical quantum yield of nanoporous carbons as multiphase systems.
The challenge on the use of sustainable metal‐free nanoporous carbon photocatalysts with enhanced response under solar light in a wide panel of technological processes is yet to design carbons with optimized features at different levels: multimodal pore control at the nanometric level, surface functionalization with chromophoric groups active and stable under sunlight, and high electron mobility. Some progress also needs to be done yet, in some key aspects toward the understanding of the fundamentals of the light/carbon/molecule interactions at a molecular level, the improvement of their photochemical quantum yields (likely by mastering the synthesis of these materials), or the stability under long illumination periods (surface photocorrosion).
Acknowledgments
This work was funded by the European Council Research through a Consolidator Grant (ERC‐CoG‐648161‐PHOROSOL).
References
- 1 Barry, T.I. and Stone, F.S. (1960). Proc. Royal Soc. 255: 124–144.
- 2 Fujishima, A. and Honda, K. (1972). Nature 238: 37–38.
- 3 Fujishima, A., Rao, T.N., and Tryk, D.A. (2000). J. Photochem. Photobiol., C 1: 1–21.
- 4 Kamat, P.V. (2014). J. Phys. Chem. Lett. 5: 4167–4168.
- 5 Serpone, N. and Pelizzetti, E. (1989). Photocatalysis: Fundamental and Applications. New York: Wiley Interscience.
- 6 Herrmann, J.M. (1999). Catal. Today 53: 115–129.
- 7 Kudo, A. and Miseki, Y. (2009). Chem. Soc. Rev. 38: 253–278.
- 8 Chang, X., Wang, T., and Gong, J. (2016). Energy Environ. Sci. 9: 2177–2196.
- 9 Li, K., An, X., Park, H.K. et al. (2014). Catal. Today 224: 3–12.
- 10 Pelaez, M., Nolan, N.T., Pillai, S.C. et al. (2012). Appl. Catal., B Environ. 125: 331–349.
- 11 Etacheria, V., Di Valentinc, C., Schneiderd, J. et al. (2015). J. Photochem. Photobiol., C 25: 1–29.
- 12 Lewerenz, H.J. (2014). J. Electrochem. Soc. 161: H3117–H3129.
- 13 Gerischer, G. and Lubke, M. (1983). Ber. Bunsen‐Ges. Phys. Chem. 87: 123–128.
- 14 Linsebigler, A.L., Guangquan, L., and Yate, J.T. (1995). Chem. Rev. 95: 735–758.
- 15 Araña, J., Doña‐Rodriguez, J.M., Tello Rendón, E. et al. (2003). Appl. Catal., B Environ. 44: 153–160.
- 16 Kumar, S.G. and Rao, K.S.R.K. (2015). Appl. Surf. Sci. 355: 939–958.
- 17 Tang, J.W., Zou, Z.G., and Ye, J.H. (2004). Angew. Chem. Int. Ed. 43: 4463–4466.
- 18 Yu, J.C., Ho, W., Yu, J. et al. (2005). Environ. Sci. Technol. 39: 1175–1179.
- 19 Litter, M.I. and Navio, J.A. (1996). J. Photochem. Photobiol., A Chem. 98: 171–181.
- 20 Minero, C., Mariella, G., Maurino, V., and Pelizzetti, E. (2000). Langmuir 16: 2632–2641.
- 21 Asahi, R., Morikawa, T., Ohwaki, T. et al. (2001). Science 293: 269–271.
- 22 Sakthivel, S. and Kisch, H. (2003). Angew. Chem. Int. Ed. 42: 4908–4911.
- 23 Cho, Y., Choi, W., Lee, C.H. et al. (2001). Environ. Sci. Technol. 35: 966–970.
- 24 Bae, E. and Choi, W. (2002). Environ. Sci. Technol. 37: 147–152.
- 25 Choi, H., Stathatos, E., and Dionysiou, D. (2006). Appl. Catal., B Environ. 63: 60–67.
- 26 Fernández, A., Lassaletta, G., Jiménez, V.M. et al. (1995). Appl. Catal., B Environ. 7: 49–63.
- 27 Leary, R. and Westwood, A. (2011). Carbon 49: 741–772.
- 28 Faria, J.L. and Wang, W. (2009). Carbon Materials for Catalysis. New York: Wiley. Chapter 13.
- 29 Matos, J., Laine, J., and Herrmann, J.M. (1998). Appl. Catal., B Environ. 18: 281–291.
- 30 Xia, X.H., Jia, Z.H., Yu, Y. et al. (2007). Carbon 45: 717–721.
- 31 Stankovich, S., Dikin, D.A., Dommett, G.H.B. et al. (2006). Nature 442: 282–286.
- 32 Wang, W., Serp, P., Kalck, P., and Faria, J.L. (2005). J. Mol. Catal., A Chem. 235: 194–199.
- 33 Ocampo‐Pérez, R., Sánchez‐Polo, M., Rivera‐Utrilla, J., and Leyva‐Ramos, R. (2011). Appl. Catal., B Environ. 104: 177–184.
- 34 Williams, G., Seger, B., and Kamat, P.V. (2008). ACS Nano 2: 1487–1491.
- 35 Tryba, B. (2008). J. Photoenergy 721824, 1–721815.
- 36 Matos, J., Laine, J., and Herrmann, J.M. (2001). J. Catal. 200: 10–20.
- 37 Velasco, L.F., Haro, M., Parmentier, J. et al. (2013). J. Catal. 178512.
- 38 Zhang, H., Lv, X., Li, Y. et al. (2010). ACS Nano 4: 380–386.
- 39 Zhang, Y., Tang, Z.R., Fu, X., and Xu, Y.J. (2010). ACS Nano 4: 7303–7314.
- 40 Luo, Y., Heng, Y., Dai, X. et al. (2009). J. Solid State Chem. 182: 2521–2525.
- 41 Ameen, S., Seo, H.‐K., Akhtar, M.S., and Shin, H.S. (2012). Chem. Eng. J. 2010: 220–228.
- 42 García, A. and Matos, J. (2010). Open Mater. Sci. J. 4: 2–4.
- 43 Matos, J., Laine, J., Herrmann, J.‐M. et al. (2007). Appl. Catal., B Environ. 70: 461–469.
- 44 Velasco, L.F., Parra, J.B., and Ania, C.O. (2010). Appl. Surf. Sci. 256: 5254–5258.
- 45 Velasco, L.F., Maurino, E., Laurenti, E., and Ania, C.O. (2013). Appl. Catal., A Gen. 453: 310–315.
- 46 Velasco, L.F., Maurino, E., Laurenti, E. et al. (2013). Appl. Catal., A Gen. 452: 1–8.
- 47 Velasco, L.F., Parra, J.B., Fonseca, I.M. et al. (2012). Carbon 50: 249–258.
- 48 Velasco, L.F., Lima, J.C., and Ania, C.O. (2014). Angew. Chem. Int. Ed. 53: 4146–4148.
- 49 Velasco, L.F., Carmona, R.J., Matos, J., and Ania, C.O. (2014). Carbon 73: 206–215.
- 50 Gomis‐Berenguer, A., Seredych, M., Lima, J.C. et al. (2016). Carbon 104: 253–259.
- 51 Velasco, L.F., Gomis‐Berenguer, A., Lima, J.C., and Ania, C.O. (2015). ChemCatChem 7: 3012–3319.
- 52 Gomis‐Berenguer, A., Iniesta, J., Maurino, V. et al. (2016). Carbon 96: 98–104.
- 53 Gomis‐Berenguer, A., Velo‐Gala, I., Rodriguez‐Castellón, E., and Ania, C.O. (2016). Molecules 21: 1611–1622.
- 54 Gomis‐Berenguer, A., Velasco, L.F., Velo‐Gala, I., and Ania, C.O. (2017). J. Colloid Interface Sci. 490: 879–901.
- 55 Velasco, L.F. (2012). Fotodegradacion oxidativa de fenol con catalizadores TiO2/C. Análisis de la respuesta fotoquímica de la fase carbonosa. PhD thesis. University of Oviedo. http://hdl.handle.net/10261/72354.
- 56 Gomis‐Berenguer, A. (2016). Photochemical response of nanoporous carbons. Role as catalysts, photoelectrodes and additives to semiconductors. PhD thesis. University of Alicante. http://hdl.handle.net/10261/151012.
- 57 Moreno‐Castilla, C. (2004). Carbon 42: 83–94.
- 58 Velasco, L.F. and Ania, C.O. (2011). Adsorption 17: 247–254.
- 59 Santos, A., Yustos, P., Quintanilla, A. et al. (2002). Appl. Catal., B Environ. 39: 97–113.
- 60 Velo‐Gala, I., López‐Peñalver, J.J., Sánchez‐Polo, M., and Rivera‐Utrilla, J. (2013). Appl. Catal., B Environ. 142–143: 694–704.
- 61 Velo‐Gala, I., López‐Peñalver, J.J., Sánchez‐Polo, M., and Rivera‐Utrilla, J. (2014). Carbon 67: 236–249.
- 62 Ania, C.O., Seredych, S., Rodríguez‐Castellón, E., and Bandosz, T.J. (2014). Carbon 79: 432–441.
- 63 Bandosz, T.J., Matos, J., Seredych, M. et al. (2012). Appl. Catal., A 445–446: 159–165.
- 64 Kuhn, H.J., Braslavsky, S.E., and Schmidt, R. (2004). Pure Appl. Chem. 76: 2105–2146.
- 65 Gimeno, O., Carbajo, M., Beltrán, F.J., and Rivas, F.J. (2005). J. Hazard. Mater., B 119: 99–108.
- 66 Rodríguez, M., Ben Abderrazik, N., Contreras, S. et al. (2002). Appl. Catal., B Environ. 37: 131–137.
- 67 Andrade, M., Mestre, A.S., Carmona, R.J. et al. (2015). Appl. Catal., A 507: 91–98.
- 68 Strelko, V.V., Kuts, V.S., and Thrower, P.A. (2000). Carbon 38: 1499–1524.
- 69 Robertson, J.J. (1992). Phys. Rev. Lett. 68: 220–223.
- 70 Oppedisano, C. and Tagliaferro, A. (1999). Appl. Phys. Lett. 75: 3650–3652.
- 71 Korver, J.A. (1950). Chem. Weekblad 46: 301–302.
- 72 Radovic, L.R., Silva‐Villalobos, A.F., Silva‐Tapia, A.B., and Vallejos‐Burgos, F. (2011). Carbon 49: 3471–3487.
- 73 Radovic, L.R. and Bockrath, B. (2005). J. Am. Chem. Soc. 127: 5917–5927.
- 74 Modestov, A.D., Gun, J., and Lev, O. (1998). Surf. Sci. 417: 311–322.
- 75 Zhu, Y., Li, X., Cai, Q. et al. (2012). J. Am. Chem. Soc. 134: 11774–11780.
- 76 Wang, A. and Chung, D.D.L. (2014). Carbon 72: 135–151.
- 77 Wypych, A., Bobowska, I., Tracz, M. et al. (2014). J. Nanomater. 2014: 124814, 1–124810.
- 78 Lee, D., Seo, J., Zhu, X. et al. (2013). Sci. Rep. 3: 2250–2255.
- 79 Beecher, J.E., Durst, T., Fréchet, J.M.J. et al. (1993). Adv. Mater. 5: 632–634.
- 80 Baca, M., Borgstahl, G.E., Boissinot, M. et al. (1994). Biochemistry 33: 143649–143677.
- 81 Finkelstein, E., Rosen, G.M., and Rauckman, E.J. (1980). Arch. Biochem. Biophys. 200: 1–16.
- 82 Yu, Y., Yu, J.C., Chan, C.Y. et al. (2005). Appl. Catal., B Environ. 61: 1–11.
- 83 Velo‐Gala, I., López‐Peñalver, J.J., Sánchez‐Polo, M., and Rivera‐Utrilla, J. (2017). Appl. Catal., B Environ. 207: 412–423.
- 84 Minero, C., Bedini, A., and Maurino, V. (2012). Appl. Catal., B Environ. 128: 135–143.
- 85 Prakash Reddy, V. and Surya Prakash, G.K. (2003). Electrophilic reactions of phenols. In: The Chemistry of Phenols (ed. Z. Rappoport), 600–660. England: Wiley.