
Biofunctionalization of Hydrogels for Engineering the Cellular Microenvironment
Maniraj Bhagawati and Sanjay Kumar, Department of Bioengineering, University of California, Berkeley, CA, USA
The in vivo cellular microenvironment is a complex network of proteins and carbohydrates that not only acts as a scaffold for cell adhesion but also provides a wide range of biochemical and physical signals that are important for directing cell behavior. This extracellular matrix (ECM) is in turn also a substrate for cell-induced degradation and remodeling. Current advances in tissue engineering and regenerative medicine have inspired the fabrication of in vitro material systems that can mimic biologically instructive properties of the ECM. In this chapter, we focus on strategies that have been developed for the functionalization of hydrogels with a wide range of biological motifs including ECM-derived proteins and growth factors. First, we consider approaches based on irreversible covalent immobilization as well as reversible and affinity-based immobilization of biomolecules onto polymer scaffolds. Next, we address approaches for the introduction of biodegradable elements such as substrates for cell-secreted enzymes into hydrogel matrices. We conclude the discussion with a short overview of techniques for synthesizing hydrogel nanoparticles, which can be used for functionalizing the surfaces of cells.
Keywords
Extracellular matrix; growth factor; hydrogel; protein immobilization; biodegradation; matrix metalloproteinase; nanoparticle
14.1 The 3D Extracellular Milieu
Tissues are composed of cells and the extracellular matrix (ECM), which provides not only physical support in the form of a scaffold but also a variety of biochemical and biomechanical signals essential for cellular function and homeostasis. The ECM is an intricate network of biopolymers that is secreted and subsequently modified, degraded, and organized by the cells adherent to it. Therefore, cells and their ECM form a closed, bidirectional loop of information transfer in which the ECM provides biochemical and mechanical signals that are conveyed from the cell membrane through the cytoskeleton to the nucleus to direct cell behavior, with cells in turn remodeling the ECM under the regulation of ECM-derived inputs [1]. Although all ECMs are nominally composed of proteins and polysaccharides, each tissue has an ECM that is distinct not only in its biochemical composition but also with respect to its physical properties, including stiffness, microstructure, and porosity. A large and still-growing body of work has demonstrated the functional importance of the ECM in regulating morphogenesis and organogenesis and mediating disease processes that occur as a result of abnormal or deregulated ECM [2–5]. Thus, the ability to fabricate in vitro cellular environments that can mimic the complex properties of the ECM holds profound value for a wide variety of applications ranging from basic cell biological studies to regenerative medicine and drug delivery.
The biochemical components of the ECM have been extensively described in several previous reviews [6–8] (Figure 14.1), and a variety of ECM proteins have been incorporated into in vitro systems for cell culture applications [9–12]. These include fibrous proteins such as collagen and fibrin, which play important roles in providing mechanical support in tissue. More specialized and tissue-specific ECM proteins, such as fibronectin (FN) and laminin, have also been included in these culture systems [13,14]. Importantly, these proteins are not merely inert scaffolds but also specifically bind cell surface adhesion receptors. The most notable among these are the integrins, which are heterodimeric transmembrane proteins that physically link the ECM and the cellular cytoskeleton [15–20]. Domain-mapping analyses of various ECM proteins have yielded the identity of several short peptide sequences that define integrin-binding specificity [21]. The best-known such sequence, and the one that has most often been included in synthetic matrices for enabling cell adhesion, is the arginine–glycine–aspartate (RGD) sequence, which was originally derived from FN repeat III10 [22]. Since then this sequence has also been found and shown to mediate cell adhesion in several other ECM proteins such as vitronectin, fibrinogen, laminin, and tenascin [21].
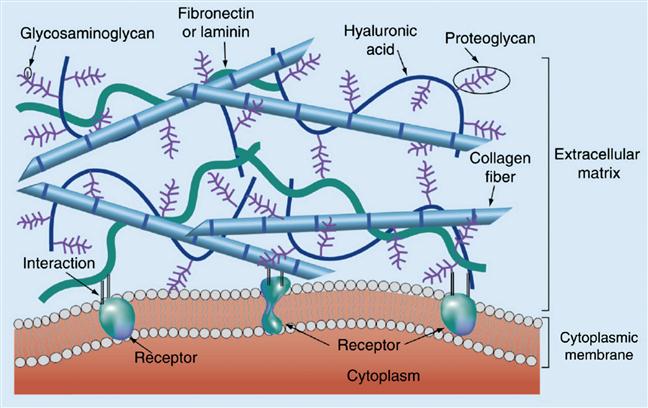
A second category of ECM macromolecules that has also been extensively used for fabrication of in vitro cell culture matrices is glycosaminoglycans (GAGs). GAGs are large linear polysaccharides consisting of disaccharide units containing an amino sugar (either GlcNAc or GalNAc) and an uronic acid (either glucuronic acid and/or iduronic acid). The amino sugar is very often sulfated, and thus GAGs are highly negatively charged polymers. GAGs, which are defined by the constituent sugar monomers and the bonds linking them together, include hyaluronans, heparan sulfates (HSs), chondroitin sulfate, dermatan sulfate, and keratan sulfate. Except for hyaluronan, all GAGs can be found covalently conjugated with proteins to form proteoglycans. In vivo, GAGs and proteoglycans perform multiple structural and mechanical roles and can trigger cell signaling by engaging cell surface receptors [23]. Another major function of GAGs, especially HS, is to locally sequester and concentrate a diverse set of growth factors and cytokines [24]. This property has inspired the use of HS (and the structurally similar molecule heparin) for enabling the entrapment of GFs in several synthetic matrices [25,26].
As alluded to earlier, the composition of the ECM is not static but is instead subject to active cell-driven remodeling, which is an important mechanism for dynamically regulating various aspects of both morphogenesis and tissue homeostasis and disease [27,28]. The major mechanism for this remodeling is degradation of the various components of the ECM by the action of cell-secreted enzymes. ECM proteins including collagens, FN, and laminin are degraded by enzymes such as matrix metalloproteinases (MMPs) [29,30] and plasmin [31]. Proteoglycans can be degraded by ADAMTS proteinases [32,33], and enzymes like hyaluronidases [34] and heparanase are specialized to degrade specific GAGs. In the context of synthetic cell culture scaffolds, cell-mediated matrix degradation can both create room for tissue growth and angiogenesis and provide a convenient mechanism for clearing the material following in vivo implantation. For these reasons, as we discuss later, there has been a significant effort to develop synthetic ECMs that are amenable to cell-mediated degradation.
14.2 Mimicking the ECM
To summarize, the ECM represents a complex, cell-interactive meshwork of proteins, proteoglycans, and GAGs that sequesters and organizes growth factors and cytokines, offers structural support to tissue, and transduces biochemical signals through specialized adhesion receptors. Thus, developing in vitro materials systems that replicate key structural and functional properties of the ECM could both facilitate the study of cell biology in tissue-like settings and provide important platforms for tissue engineering, drug discovery, and delivery of therapeutic cells and proteins in vivo [35–41]. Hydrogels, which are water-swollen, 3D networks of polymers, have demonstrated great potential to accomplish these goals. Hydrogel networks can be structurally stabilized by a number of mechanisms such as covalent cross-linking, physical entanglement, formation of crystallites, and assembly through a variety of noncovalent interactions including hydrogen bonding and electrostatic forces [42,43]. The highly water-swollen nature of hydrogels confers important mechanical properties and facilitates the encapsulation of cells. Hydrogel materials often exhibit strong biocompatibility and high permeability for oxygen, nutrients, and other water-soluble metabolites [44–49].
Hydrogels have been fabricated from both natural and synthetic polymers. Natural polymers are typically biocompatible, inherently biodegradable, and often contain motifs that direct specific biological functions. ECM-derived proteins such as elastin, fibrin, collagen, and the collagen derivative gelatin have been traditionally used to form hydrogels [50–53]. Despite their advantages, ECM proteins derived from tissue can suffer from concerns over immunogenicity, retention of infectious agents, poor scalability, and high batch-to-batch variability. Furthermore, hydrogels based on natural polymers offer limited opportunity for bottom-up design of desired physicochemical properties or biological functionalities. For these reasons, hydrogels based on recombinantly expressed proteins have recently emerged as a complementary class of biological matrices. One of the most well-known examples of such systems is inspired from recurring amino acid sequences found in tropoelastin and are thus known as elastin-like polypeptides (ELPs) [54]. ELP-based hydrogels have proved promising for both experimentally modeling and serving as a therapeutic platform for a wide variety of tissues including nerve, skeletal muscle, and cartilage [55–59]. Peptide hydrogels have also been fabricated using amphiphilic diblock copeptides [60,61] and peptides that fold to form amphiphilic β-hairpins that subsequently self-assemble into a hydrogel network [62–64]. Advances in protein- and peptide-based hydrogels have been covered in several recent reviews [65,66].
Tissue-derived GAGs and a variety of non-ECM polysaccharides have also been extensively used for fabricating hydrogels. These polysaccharides can be modified with diverse functional groups such as acrylates, thiols, and amines to allow crosslinking. Hyaluronic acid (HA), which is ubiquitous in the ECM of many tissues, has been extensively used to fabricate hydrogels for application in a wide range of study systems [67,68]. Since HA levels have been shown to be elevated in the stroma of cancers [69,70], hydrogels based on HA have been used for modeling the tumor microenvironment in breast, colon, ovarian, and lung cancers [71–73]. HA also forms a large fraction of the ECM in brain, and so HA-based hydrogels have also been used to model brain ECM in vitro. Such hydrogels have found particular use in the study of the brain tumor glioblastoma multiforme [74,75]. Other polysaccharides that have been utilized for fabricating hydrogels include heparin, chitosan, dextran, and alginate [76–80].
Synthetic polymer hydrogel systems have a long history of use in medical devices, implants, and other materials, and remain of significant interest for use as ECM scaffolds. In general, synthetic polymers offer greater batch-to-batch consistency and significantly more physicochemical tailorability than native biological materials. For example, poly(ethyleneglycol) (PEG) is perhaps the most widely used synthetic polymer hydrogel system. PEG has several unique properties such as high solubility in water and various organic solvents, low intrinsic toxicity and immunogenicity, and comparatively low nonspecific protein adsorption [81–85]. In addition, the hydroxyl end groups of PEG are easily modified with other chemical functionalities that can be used for crosslinking as well as for functionalization with a variety of cell-reactive moieties. While a detailed discussion of all synthetic polymer systems currently under use for hydrogel fabrication is beyond the scope of this chapter, several extensive reviews have been written on this subject [44,86,87].
While native, recombinant, and synthetic hydrogels have their relative advantages and disadvantages, many of these systems share the need for chemical “postprocessing” to achieve maximal biological function. For example, whereas native protein hydrogel systems such as those based on collagen and fibrin encode a rich palette of biological instruction, they may require chemical crosslinking to achieve the desired mechanical properties for a given application or conjugation of growth factors to promote specific phenotypes. Conversely, whereas a PEG hydrogel may have ideal physical properties for a given biological application, its bio-inert nature requires all adhesive and biodegradation properties to be built into the system, typically by attachment of proteins and peptides to the polymer backbone. We now discuss these conjugation strategies (Figure 14.2) in greater detail.
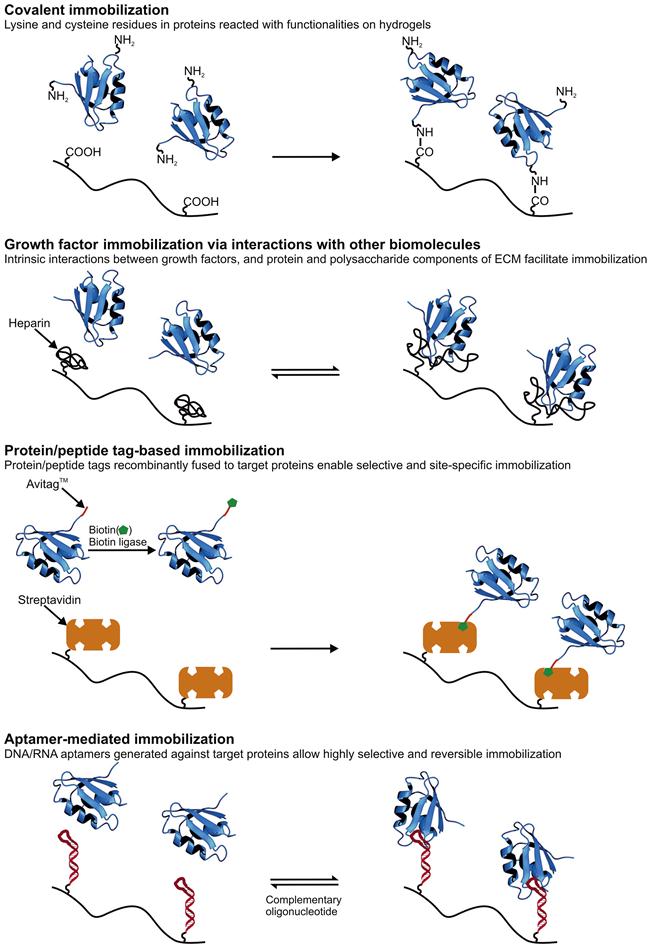
14.2.1 Covalent Immobilization of ECM-Derived Proteins and Growth Factors Through Reaction of Amino Acid Side Chains
The side chains of several amino acids offer chemically reactive handles for bioconjugation. These include the amine group of lysine, the thiol group of cysteine, and the carboxylic acid groups of aspartic and glutamic acid. With the development of orthogonal genetic codes and synthetic tRNA systems that enable the expression of recombinant proteins containing unnatural amino acids, the reactive functionalities available on proteins have expanded even further [88–90]. Therefore, covalent coupling of these functionalities with reactive groups on hydrogels or hydrogel precursors is one of the most heavily utilized strategies for immobilization of proteins and peptides on hydrogels. Full-length FN, laminin, and collagen, as well as adhesive peptide motifs derived from these and other proteins have been incorporated into hydrogels using this strategy [91–93]. Growth factors and derived peptide motifs have also been immobilized on a variety of matrices using covalent reaction with lysine and cysteine residues (Figure 14.2) [94,95].
Lysines can be directly conjugated to scaffold carboxylic acids or their N-hydroxysuccinimide (NHS) derivatives leading to the formation of stable amide bonds. In this strategy, the carboxylic acid has to be incorporated into the hydrogel prior to protein immobilization. If the polymer does not already contain carboxylate functionality, it can be introduced either by copolymerizing carboxylic acid containing monomers with the polymer backbone or by post hoc carboxylate functionalization of the assembled hydrogel. The first strategy has proved valuable for modifying acrylamide-based hydrogels copolymerized with NHS-ester moieties with ECM proteins such as FN, vitronectin, and collagen [91]. Similarly, copolymerization of acrylic acid with PEG-diacrylate (PEGDA) has facilitated the formation of hydrogels which could be efficiently functionalized with several short peptide motifs derived from FN, laminin, and collagen [92]. The carboxylic acid groups already present in HA have also been used to immobilize rat tail collagen I [96]. As examples of the second strategy, poly(vinylalcohol) (PVA) hydrogels have been carboxylate functionalized with 11-bromoundecanoicacid, activated to yield NHS-esters, and subsequently conjugated with FN [97]. The amine-reactive photo-linkable crosslinker sulfo-SANPAH has also been widely used to activate hydrogels, especially defined stiffness polyacrylamide hydrogels, for immobilization of ECM proteins through lysine residues [96,98]. Sulfo-SANPAH has also been used to conjugate laminin to methylcellulose hydrogels [99].
As an alternative to directly reacting the amine groups of the target protein to the polymer backbone, these amines can instead be conjugated to a small molecule that can react/interact with chemical functionalities available on the polymer backbone. For example, proteins have frequently been modified with acrylate groups, which allow these proteins to be co-photopolymerized with synthetic polymers containing acrylate groups to create bioactive hydrogels. This strategy has been used to conjugate FN to hydrogels made from PEG [100] and HA [101], thus demonstrating the flexibility of its application. Acryl-PEG-NHS has also been used to modify integrin-binding peptides such as RGD and YIGSR [102,103], as well as a variety of growth factors including fibroblast growth factor-2 (FGF-2, also known as basic FGF) [94] and platelet-derived growth factor-BB (PDGF-BB) [104], in order to facilitate the incorporation of these bioactive species into PEGDA hydrogels. Similarly, lysine residues in epidermal growth factor (EGF) have been functionalized with iodoacetamide-NHS to enable conjugation of this growth factor on HA hydrogels carrying thiol groups [105]. Another secondary functionality that enables high-affinity noncovalent conjugation is biotin, which can be used to link the modified biomolecule to avidin-functionalized hydrogels [106–108].
Although the abundance of lysines in proteins makes lysine an attractive residue to use for immobilization, it is challenging if not impossible to precisely control which lysine (or lysines) in a given protein reacts with the hydrogel matrix. This leads to an inhomogeneous distribution of protein orientation, which could lead to some fraction of the immobilized protein molecules to be denatured or sterically shielded from their interaction partners on the cell surface. Furthermore, conjugation of functionally critical lysine residues could compromise or eliminate the bioactivity of the protein. Therefore, cysteine residues, which are significantly rarer in proteins, can be used for more selective immobilization. Using recombinant DNA technology, cysteines can also be introduced into target proteins at precisely specified positions along the polypeptide chain, which enables a high degree of control over the orientation of immobilization. The glutathione S-transferase fusion tag which is often used for affinity purification of recombinantly expressed proteins also contains cysteine residues that have been used for immobilization on thiol-reactive matrices [109].
Thiols from cysteine residues can take part in several reactions that have been demonstrated to be very useful for biofunctionalization of hydrogels. One of the most widely used strategies is the Michael addition reaction in which a thiol reacts with an alkene, often in the context of an acrylate or methacrylate moiety. This chemistry has been used to attach FN to PEGDA, which was then photo-crosslinked to yield a hydrogel network [109]. A similar scheme has also been used to first generate FN-functionalized PEGDA and then crosslink thiolated-HA into the PEG network to create a three-component hydrogel [93]. A similar strategy has also been used to fabricate injectable HA hydrogels decorated with RGD peptide for the in vivo delivery of encapsulated cells [110]. In this study, fibroblasts were added to a mixture of thiolated-HA and RGD-modified PEGDA. The resulting mixture was then subcutaneously injected into the flanks of nude mice, leading to the formation of hydrogels in vivo. In a strategy employing the same chemistry but in an opposite topology, HA has been methacrylated using methacrylic anhydride and subsequently reacted with RGD peptides containing a cysteine residue [75,111]. PEGDA has also been conjugated with a thiolated transforming growth factor-β (TGF-β), which enabled the fabrication of TGF-β-functionalized PEG hydrogels [112]. Vinylsulfone moieties, which react more selectively and rapidly with thiols [113], have also been used for functionalization of hydrogels. PEG macromers carrying vinylsulfone terminal groups have been reacted with cysteine-terminated RGD and other peptides to introduce integrin-binding sites in hydrogels [114]. The same reaction has also been utilized to incorporate vascular endothelial growth factor (VEGF) carrying a genetically engineered cysteine residue into PEG hydrogels [115]. Thiol groups can also react with maleimides in a variation of the Michael addition reaction which proceeds with significantly faster kinetics [116]. This reaction has been extensively used for immobilizing peptides derived from ECM proteins onto PEG-based hydrogels [117]. Maleimide-terminated multiarm PEGs (often referred to as star-PEGs) have been modified with RGD and FN through cysteine residues. PEG arms with unreacted maleimide groups could be subsequently reacted with thiol modified PEGs thus crosslinking the polymers into a hydrogel [118]. Heparin functionalized with maleimide groups could also be incorporated into such hydrogels to enhance the bioactivity of the hydrogel system and endow it with growth-factor-binding properties [24]. PEG-maleimide has also been reacted with VEGF in order to allow its incorporation in PEG hydrogels [95]. Another fast and thiol-specific reaction that may be carried out in the presence of living cells is the photoactivated thiolene reaction [119], which has been utilized for immobilization of FN- and laminin-derived peptides on PEG hydrogels [120]. A major advantage of this technique over the earlier strategies is its photosensitive nature, which makes it amenable for spatial patterning of peptides and proteins on hydrogels. Indeed, 3D micropatterning of RGD peptides on PEG-based hydrogels has been achieved using this chemistry [121]. In an elegant approach utilizing click chemistry for hydrogel crosslinking and the thiolene reaction for peptide immobilization, PEG hydrogels with independently controllable mechanical and biochemical parameters could be fabricated [122].
Use of reactive amine and thiol groups already present in the protein sequence is a very attractive approach due to its flexibility and ease of application. However, in addition to the limitations described earlier, a shortcoming of these strategies is their lack of selectivity against cell- and medium-derived proteins, which might themselves couple to the polymer backbone. In addition to compromising yield by consuming reactive sites, this could confound studies that seek to delineate the effect of specific ECM proteins or derived peptides upon cellular processes. Therefore, several methods have been established that enable the selective immobilization of target proteins of interest onto hydrogel platforms, some of which are covered below.
14.2.2 Immobilization of Growth Factors Through Interactions with Other Biomolecules
Growth factors interact with several components of the ECM. In this section we describe ECM proteins and GAGs that exhibit such interactions and hydrogel systems that have taken advantage of some of these interactions to entrap cell-derived factors (Figure 14.2).
As described earlier, the interactions between GAGs and growth factors have been extensively characterized and leveraged for biomaterial design. GAG–protein interactions are largely based on electrostatic attraction between cationic residues on the proteins and anionic sulfate and carboxyl groups on the GAGs, although hydrogen bonding and van der Waals interaction are also believed to contribute [123,124]. Through these interactions GAGs are thought to sequester growth factors, providing a depot effect that regulates these proteins’ local concentration, diffusion, and signaling intensity [125]. Moreover, the association of growth factors with heparin and HS has been shown to increase growth factor lifetime by protecting these proteins from enzymatic degradation [126,127].
Among the GAGs, heparin and HS are probably the most extensively studied molecules based on their interaction with diverse growth factors such as FGFs, TGF-β, VEGF, and PDGF-A and -B [24] (Table 14.1). FGFs are among the best-studied heparin-binding proteins, and structural studies have shown that heparin and HS interact with both FGF and its receptor, while simultaneously promoting receptor dimerization [128,129]. Hydrogels based on heparin and HS have therefore found extensive use as platforms for sustained release of growth factors. A heparin-crosslinked PEG-based hydrogel loaded with VEGF demonstrated gradual release of active VEGF over a period of 3 weeks [130]. Subcutaneous implantation of these hydrogels in mice was shown to induce angiogenesis around the implantation area. Taking this a step further, a similar strategy was used to enable the simultaneous sustained delivery of VEGF and FGF-2, which demonstrated superior pro-angiogenic activity over administration of single growth factors in both in vitro and in vivo models [131]. PEG-heparin hybrid hydrogels loaded with hepatocyte growth factor (HGF) were shown to not only support the culture of primary rat hepatocytes but also promote urea and albumin synthesis, which has motivated the application of this platform for in vitro differentiation of hepatocytes and as vehicles for transplantation [132]. Heparin has also been combined with natural polymers in order to synthesize sustained growth factor-releasing hydrogels. In a related study, heparin was incorporated into chemically crosslinked alginate hydrogels and used to entrap FGF-2 [133]. Here, regenerated axons from rat sciatic nerves grew much faster on heparin-alginate hydrogels than on alginate-only hydrogels, suggesting that heparin promoted retention and delivery of the growth factor. In another study, heparin was incorporated into photo-crosslinked alginate hydrogels and the hydrogels were loaded with bone morphogenetic protein-2 (BMP-2). It was demonstrated that BMP-2-loaded photo-crosslinked heparin-alginate hydrogels induced significantly more osteogenesis than unmodified alginate hydrogels loaded with BMP-2 [25]. Hydrogels formed from HA have also been functionalized with heparin in order to achieve controlled release of BMP-2 [26]. GAGs other than heparin are known to interact with specific growth factors [134–136], but these materials have not yet been as extensively explored as matrices for controlled release.
Table 14.1
Interactions Between Growth Factors and ECM Components
| ECM Component | Interacting GF |
| Heparin/heparan sulfate | FGFs [24,128], TGF-βs [283], VEGF [284,285], PDGFs [286,287], HGF [136,288], BMPs [289–291] |
| Collagens | PDGFs [151], HGF [152], TGF-β1 [153] |
| Vitronectin | IGF-II [154], TGF-β1 and 2, EGF, VEGF, FGF-2 [156], HGF [141] |
| Fibronectin | HGF [141], TGF-β1 [140], VEGF [137,144,145], CTGF [142], FGFs [137,146], PDGFs [137,143], PIGF-2, 3, BMP-2, 7, NGF, BDNF [137] |
| Fibrinogen | VEGF [138,149], FGFs [138,148], PIGF-2,3, PDGFs, BMP-2,2/7, TGF-β1,2, BDNF [138] |
| Tenascin-C | PDGFs, VEGF, PIGF-2,3, FGFs, TGF-β1,2, BMP-2, BDNF, HGF [150] |
Growth factors have also been shown to interact with ECM protein components. Recently, several ECM proteins such as FN, fibrinogen, and tenascin-C were shown to bind a large number of diverse growth factors (Table 14.1) [137,138]. The regions of these proteins responsible for binding growth factors were localized to their heparin-binding motifs, although binding surprisingly does not require the presence of heparin. Interestingly, mutation of the positively charged residues in these motifs into serines abolishes growth factor binding indicating that the interactions may be driven by electrostatic forces.
FN is an important component of the ECM in wounds and its interaction with growth factors is believed to be important for wound healing [139]. FN is known to interact with TGF-β1, HGF, connective tissue growth factor (CTGF), PDGF-A, and VEGF with high affinity [140–144]. Using recombinant FN domains, the C-terminal heparin-binding Hep-II domain of FN (consisting of type III repeats 13 to 14) was identified as the key site for binding VEGF and FGF-2 [145,146]. Interestingly, peptide fragments containing both the α5β1 integrin-binding domain (FN type III repeats 9 to 10) and VEGF-binding Hep-II domain significantly enhanced VEGF-induced cell responses, suggesting strong crosstalk between bound integrins and the VEGF receptor mediated by FN and possibly by other ECM-derived proteins [145]. In a high-throughput study, it was observed that the heparin-binding FN III12-14 domain binds most of the growth factors from the PDGF, VEGF, and FGF families, and some growth factors from the TGF-β and neurotrophin families with nanomolar affinity and without inhibiting growth factor activity [137]. A total of 25 previously unknown interactions with growth factors were identified in this report. Growth factor binding was enhanced in the presence of heparin, but the extent of enhancement was variable.
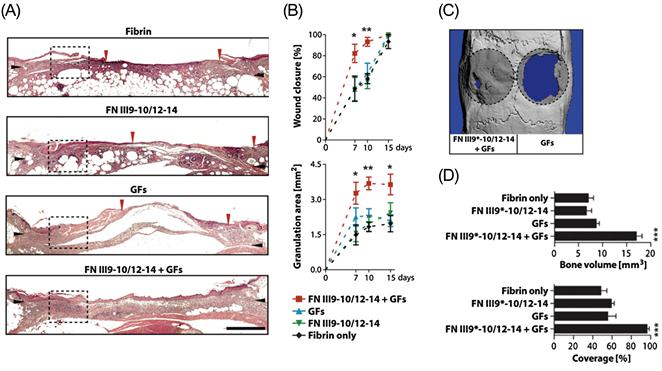
The therapeutic implications of this discovery were clearly demonstrated in a study in which the multiple interactions of this domain were utilized to sequester a variety of growth factors in a fibrin-based hydrogel [147]. In this study, a fusion peptide consisting of the integrin-binding FN III9-10 domain and the growth factor-binding FN III12-14 domain previously shown to have a synergistic effect on enhancing VEGF-induced cell signaling [145] was used to functionalize fibrin hydrogels. These hydrogels were shown to sequester VEGF-A165, PDGF-BB, and BMP-2, and enhanced the morphogenetic effects of these growth factors in vitro. Significantly, it was further demonstrated that the FN III9-10/12-14 peptide greatly enhanced the regenerative effects of the growth factors in vivo in a diabetic mouse model of chronic wounds and in a rat model of critical-size bone defects at doses where the growth factors delivered within fibrin only had no significant effect (Figure 14.3).
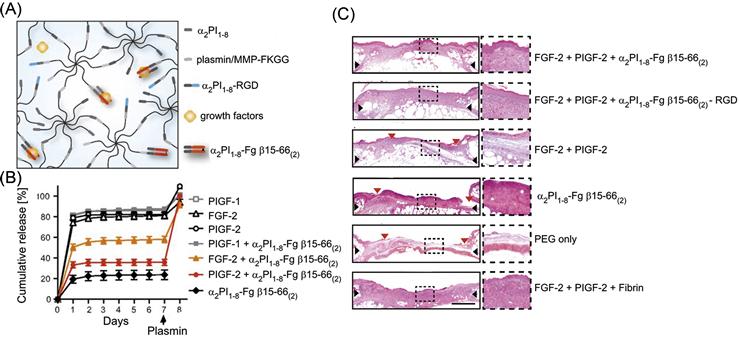
Another protein system that has been extensively studied for its interactions with growth factors is fibrinogen and its enzymatic degradation product fibrin. Fibrinogen is found in blood plasma and plays an important function in clotting by acting as a scaffold for platelet adhesion and by promoting endothelial cell mitogenesis [139]. FGF-2, which is released by disrupted blood vessels during wound formation, can bind both fibrin and fibrinogen [148], and this sequestration is necessary for endothelial cell migration, mitogenesis, and ultimately angiogenesis during wound repair. VEGF, another growth factor involved in wound healing, also interacts with both fibrinogen and fibrin [149]. A recent high-throughput study identified 15 unique interactions between the ECM proteins fibrin and fibrinogen and growth factors from the PDGF, VEGF, FGF, TGF-β, and neurotrophin families [138]. This study then demonstrated that similar binding characteristics are shown by a dimer of the heparin-binding domain of fibrinogen. Significantly, binding with this domain did not alter the ability of growth factors to trigger cell proliferation. The researchers then incorporated this domain into a synthetic PEG-based hydrogel and showed that the domain effectively sequestered FGF-2 and placental growth factor-2 (PlGF-2) (Figure 14.4). Furthermore, this synthetic bioactive hydrogel could fully recapitulate the efficacy of a fibrin matrix in promoting wound healing in a mouse model.
Similar promiscuous growth factor-binding behavior was recently demonstrated for the heparin-binding domain of Tenascin-C (TNC) [150]. In this study, recombinant peptide fragments of TNC representing the first five TNCIII repeats (TNCIII1–5) were used to study interactions with various growth factors. Multiple growth factors of the PDGF, FGF, and TGF-β families were found to interact with this region of TNC, specifically to TNCIII5.
Collagens I–VI have been shown to bind PDGF-AA, BB, and AB with high affinity without affecting the activity of these growth factors [151]. Collagens can also bind HGF with nanomolar affinity, and HGF bound to collagen has been shown to retain its pro-motility and proliferative functions in culture [152]. Similarly, collagen was also shown to bind TGF-β [153].
Finally, insulin-like growth factor-II (IGF-II) is known to interact with vitronectin [154]. It was shown that IGF-II bound to vitronectin significantly enhanced migration of breast cancer cells in transwell assays, while unbound IGF-II did not [155]. Vitronectin is also known to interact with a variety of other growth factors such as TGF-β, EGF, FGF-2, VEGF, and HGF [141,156].
Thus, intrinsic interactions between growth factors and various components of the ECM can be utilized to entrap these factors in hydrogels based on both natural and synthetic polymers, with retention of biological activity. Furthermore, the typically reversible nature of the scaffold–factor interaction makes such strategies very attractive for therapeutic applications requiring sustained in vivo delivery of growth factors. A major shortcoming of such approaches is that they are not universally generalizable but are instead limited to specific growth factors with high affinity for a given scaffold. The following sections describe some strategies that can be generically applied for the immobilization of any target protein on hydrogels.
14.2.3 Protein/Peptide Tags for Immobilization
Recombinant DNA technology has been used to tag growth factors and ECM-derived proteins with peptides that enable their specific immobilization onto hydrogels (Figure 14.2). In this way, amino acids in the native sequence that may be critical to function are spared from chemical functionalization. Furthermore, placement of the tag in a known position relative to the bioactive portion(s) of the protein increases the likelihood that protein orientation is homogeneous. Such strategies are also more generic in that most proteins can be recombinantly fused to such tags without affecting their native function as well as expressed in material-scale quantities using bacterial, yeast, and insect cell expression systems.
14.2.3.1 Collagen-Binding Domain-Mediated Immobilization
One of the most common families of peptide tags that have been used for targeted protein immobilization on hydrogels are derived from the collagen-binding domains (CBDs) of proteins. Such domains have been obtained from four sources—the von Willebrand factor (vWF), mammalian collagenase, bacterial collagenase, and FN [157].
The CBDs derived from vWF and human collagenase are ten and seven residues long, respectively. Due to their short lengths, these peptides are preferred fusion tags for growth factors in order to impart them with collagen-binding properties. The CBD derived from vWF has been fused to several growth factors such as TGF-β [158,159], EGF [160], FGF-2 [161], BMP-2 [162,163], and BMP-3 [164]. In vitro experiments demonstrated that collagen matrices functionalized with the TGF-β-CBD fusion protein, but not commercial TGF-β, could promote the survival, proliferation, differentiation, and colony mineralization of osteogenic precursor cells present in rat bone marrow [159]. In an in vivo rat ectopic bone formation assay, implantation of collagen hydrogels functionalized with BMP-2 fused to this CBD induced the production of new bone tissue inside the implants [163]. These tissues displayed the hallmarks of mature bone such as trabecular morphology and medullary cavities. CBD-BMP-3 fusion proteins affixed to collagen hydrogels also stimulated ingrowth of new bone in a rat cranial defect model, underlining the high therapeutic potential of such growth factor fusion constructs [164]. The CBD derived from human collagenase has also been fused to a large number of growth factors in order to endow them with collagen-binding properties. Both in vitro and in vivo experiments using collagen scaffolds functionalized with growth factors such as PDGF-BB [165,166], nerve growth factor-β (NGF-β) [167], neurotrophin-3 [168], VEGF [169,170], EGF [171], and FGF-2 [172] fused to this CBD have demonstrated the great potential of this strategy for tissue engineering and medical applications. A comparison of the fusion tags derived from vWF and human collagenase showed that the latter enhanced collagen affinity. This also translated to in vivo activity, as collagen scaffolds functionalized with FGF-2 fused to the collagenase-derived peptide were able to stimulate greater vascularization upon subcutaneous implantation in rats [172]. The CBD of bacterial collagenase and FN are comparatively larger proteins (24 and 27 kDa respectively) and can thus affect the functionality of growth factors fused to them. Despite this, the bacterial collagenase-derived domain has been fused with FGF-1 [173,174], FGF-2 [175], EGF [175], and VEGF [176], while the CBD of FN has been combined with HGF [177], BMP-4 [178], EGF [179], and VEGF [180] to endow these growth factors with collagen-binding ability. The BMP-4 fusion protein was strongly osteoinductive in 3D cultures of human bone-marrow-derived mesenchymal stem cells in a hybrid scaffold based on collagen and poly(lactic-co-glycolic acid) [178]. In vivo implantation of such scaffolds demonstrated that the immobilized CBD-BMP4 retained its osteoinductive activity, as evidenced by its ability to upregulate osteogenic gene expression and biomineralization.
14.2.3.2 Factor XIIIA Transglutaminase Catalyzed Immobilization
During wound healing, fibrin clots are stabilized by the transglutaminase factor XIIIA (FXIIIa), which crosslinks glutamine residues on one fibrin molecule to lysine residues on another fibrin molecule through isopeptide bonds. This reaction is also used in the in vitro fabrication of fibrin hydrogels and has been extended to the conjugation of target proteins and peptides to such hydrogels [181–183]. This strategy is based on a peptide derived from the 12 N-terminal residues of α2-plasmin inhibitor (α2-PI), which was found to be a good substrate for FXIIIa and could be readily crosslinked with fibrin via the transglutamination reaction catalyzed by FXIIIa [184]. Thus, proteins or peptides fused to this FXIIIA substrate peptide could be covalently conjugated onto fibrin matrices as demonstrated for two integrin-binding peptides FN-derived RGD and collagen-derived DGEA [185]. This α2-PI-derived peptide domain was also fused with heparin biding peptides, which were then immobilized on fibrin scaffolds in order to enable the incorporation of heparin into the hydrogels [181]. This strategy was then utilized to sequester the heparin interacting growth factor, FGF-2. It was demonstrated that FGF-2 immobilized within heparin-modified fibrin enhanced neurite extension by up to 100% relative to unmodified fibrin [182]. The promiscuous growth factor-binding domain of FN was also immobilized onto fibrin hydrogels using this reaction [137,147].
Growth factors can also be recombinantly fused to the α2-PI-derived peptide sequence to allow immobilization onto fibrin matrices (Table 14.2). This was first demonstrated for NGF-β [186]. A peptide sequence that could be cleaved by the cell-secreted protease plasmin was included between the α2-PI-derived peptide and NGF-β to ensure that the growth factor, which was irreversibly immobilized onto the matrix, would be released and be able to stimulate signaling only in the vicinity of plasmin-secreting invading cells. This NGF fusion protein immobilized onto a fibrin matrix enhanced neurite extension from embryonic chick dorsal root ganglia by 50% relative to soluble NGF. Other growth factors that have been recombinantly fused to the α2-PI-derived peptide in order to enable incorporation into fibrin hydrogels include VEGF [183,187], BMP-2 [188], and IGF-1 [189]. For growth factors not amenable to recombinant expression, the peptide sequence can also be covalently reacted with surface lysines as has been done for keratinocyte growth factor [190].
Table 14.2
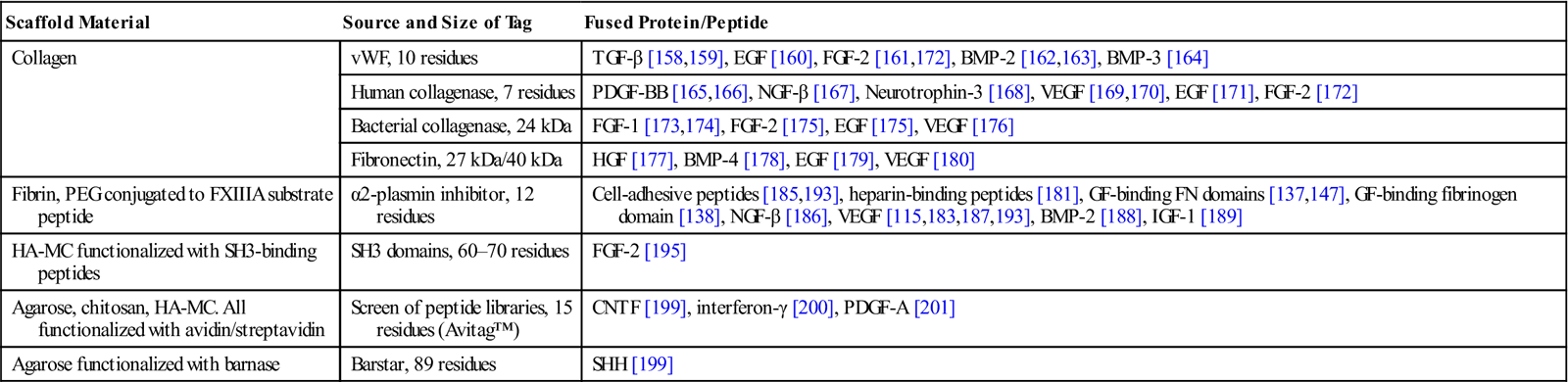
Protein and Peptide Tags That Can Be Used for Site-Specific Immobilization of Target Fusion Proteins/Peptides
| Scaffold Material | Source and Size of Tag | Fused Protein/Peptide |
| Collagen | vWF, 10 residues | TGF-β [158,159], EGF [160], FGF-2 [161,172], BMP-2 [162,163], BMP-3 [164] |
| Human collagenase, 7 residues | PDGF-BB [165,166], NGF-β [167], Neurotrophin-3 [168], VEGF [169,170], EGF [171], FGF-2 [172] | |
| Bacterial collagenase, 24 kDa | FGF-1 [173,174], FGF-2 [175], EGF [175], VEGF [176] | |
| Fibronectin, 27 kDa/40 kDa | HGF [177], BMP-4 [178], EGF [179], VEGF [180] | |
| Fibrin, PEG conjugated to FXIIIA substrate peptide | α2-plasmin inhibitor, 12 residues | Cell-adhesive peptides [185,193], heparin-binding peptides [181], GF-binding FN domains [137,147], GF-binding fibrinogen domain [138], NGF-β [186], VEGF [115,183,187,193], BMP-2 [188], IGF-1 [189] |
| HA-MC functionalized with SH3-binding peptides | SH3 domains, 60–70 residues | FGF-2 [195] |
| Agarose, chitosan, HA-MC. All functionalized with avidin/streptavidin | Screen of peptide libraries, 15 residues (Avitag™) | CNTF [199], interferon-γ [200], PDGF-A [201] |
| Agarose functionalized with barnase | Barstar, 89 residues | SHH [199] |
Notes: (I) Not all listed proteins with CBD fusion have been immobilized on hydrogels. Relevant proteins that have been successfully expressed and purified in bioactive collagen-binding forms have been included and are expected to bind to collagen scaffolds.
(II) The matrix material is not limiting for the other tags as long as they are modified with the interacting peptide/protein tag.
Interestingly, this enzymatic immobilization strategy has been now extended to enable functionalization of synthetic PEG-based matrices as well (Table 14.2). In this approach, multiarm PEG molecules are functionalized with either the α2-PI-derived peptide (acceptor peptide) or with the sequence FKGG (donor peptide), which was previously optimized as an efficient substrate for the transglutamination reaction catalyzed by FXIIIa [191]. Cross-linking of these two PEG formulations by FXIIIa yields a hydrogel, with inclusion of peptides or proteins fused to the acceptor peptide sequence during the crosslinking reaction incorporating these sequences into the hydrogel. Using this strategy, peptides containing the RGD sequence were immobilized onto PEG hydrogels which allowed mammalian cells to adhere and proliferate on the hydrogels [192]. The heparin-binding domain of fibrinogen, which is known to bind several growth factors, was also incorporated into PEG hydrogels using this technique in order to get fibrin-mimetic synthetic hydrogels which efficiently retained growth factors that showed interactions with the fibrinogen domain [138] (Figure 14.4). Such hydrogels could be efficiently loaded with the pro-angiogenic growth factors, FGF-2 and PIGF-2, and could fully mimic the effect of fibrin in promoting wound repair in a diabetic mouse model of impaired wound healing. Recombinant VEGF was also quantitatively incorporated into PEG-based hydrogels using this strategy [193].
14.2.3.3 Src Homology 3 Domain-Mediated Immobilization
Src homology 3 (SH3) domains are independently folding 60–70-residue peptide domains that bind polyproline sequences in proteins [194]. This interaction was used to immobilize FGF-2 fused to an SH3 domain on hyaluronic acid-methyl cellulose (HA-MC) composite hydrogels functionalized with SH3-binding polyproline peptide sequences [195]. Notably, this study also showed that hydrogels functionalized with two different polyproline peptide sequences having distinct affinities to the SH3 domain showed markedly different release profiles of FGF-2, which demonstrated the utility of this strategy to enable tunable release of growth factors and other protein drugs for therapeutic applications.
14.2.3.4 Enzymatic Biotinylation of Proteins for Immobilization
The Escherichia coli biotin ligase BirA catalyzes the transfer of biotin to the amino group of a lysine residue of the biotin carboxyl carrier protein subunit of acetyl-CoA carboxylase. Screening of peptide libraries has led to the discovery of a 14-residue peptide sequence that can be biotinylated by BirA at rates similar to the native substrate [196]. This sequence has since been modified by the addition of a C-terminal glutamate residue and termed the Avitag™ system. Proteins recombinantly fused to this peptide can be site-specifically biotinylated and then modified with streptavidin. This strategy has been used for conjugating recombinant proteins with fluorescent probes [197] and nanoparticles [198]. Recently, this enzymatic reaction was utilized for immobilization of recombinantly expressed growth factors and cytokines on hydrogels as well. In one report, thiolated-agarose hydrogels were first reacted with maleimide-functionalized streptavidin, which was subsequently used to capture ciliary neurotrophic factor (CNTF) fused to the AviTag that was biotinylated prior to immobilization [199]. In another paper, the pro-neuronal differentiation factor interferon-γ was recombinantly fused to the AviTag™ and immobilized on streptavidin-modified chitosan hydrogels [200]. Neural stem/progenitor cells (NSPCs) cultured on these matrices showed preferential differentiation into neurons confirming the retention of bioactivity of the cytokine even after immobilization. In order to promote differentiation of NSPCs into oligodendrocytes, the oligodendrocyte-differentiating factor PDGF-A was biotinylated using this reaction and then immobilized on hybrid HA-MC hydrogels that were previously modified with streptavidin [201]. As expected, rat NSPCs differentiated into significantly more oligodendrocytes when cultured in such hydrogels.
14.2.3.5 Barstar-Mediated Immobilization
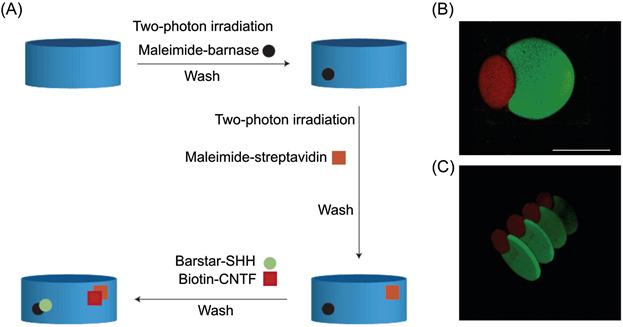
The bacterial ribonuclease barnase and its inhibitor barstar bind each other with extremely high affinity [202,203]. This strong binding has been utilized to immobilize the stem cell differentiation factor sonic hedgehog (SHH) recombinantly fused to barstar on barnase-functionalized hydrogels [199]. In this work, photosensitive caging of thiol groups on agarose hydrogels was used to pattern SHH and CNTF with a very high spatial resolution. Here, two-photon irradiation was used to uncage thiol groups in selected regions of the hydrogel followed by immobilization of barnase modified with maleimide. Next, thiol groups in another region were uncaged and reacted with streptavidin conjugated to maleimide groups. Upon immersing the hydrogel in a solution of SHH-barstar and biotin-CNTF, the proteins were independently immobilized into the target regions (Figure 14.5). The high binding affinity of both interactions ensured that binding was almost irreversible. Mouse retinal precursor cells cultured in these matrices expressed downstream effectors of both SHH and CNTF signaling pathways, indicating that the immobilized proteins retained their activity.
14.2.4 Aptamer-Mediated Immobilization
Aptamers are single stranded DNA or RNA sequences that bind with target proteins with a specificity and affinity comparable to that of antibodies [204]. These sequences are most often selected in vitro from a pool of random oligonucleotides, and selection conditions can be manipulated to obtain aptamers with different specificities and affinities [205]. Aptamers have been identified that bind a variety of proteins including ECM components [206–209] and growth factors [210–225] (Table 14.3).
Table 14.3
Growth Factor, Cytokine, and ECM Protein Targets of Aptamers
| Target | Affinity (nM) |
| VEGF [216] | 0.14 |
| PDGF-AB, PDGF-BB [210] | 0.1 |
| FGF-2 [212] | 0.13 |
| IGF-II [217] | 2 |
| TGF-β1 [218] | 90 |
| TGF-β2 [219] | 5 |
| KGF [214] | 0.0003 |
| Angiopoietin-1 [220] | 2.8 |
| Angiopoietin-2 [221] | 2.2 |
| Erythropoietin-α [225] | 33 |
| TNF-α [223] | 7 |
| Interferon-γ [224] | 1.8 |
| Interleukin-17A [225] | 0.05 |
| Tenascin-C [208] | 5 |
| Osteopontin [209] | 18 |
| Fibronectin [206] | Not reported |
Taking advantage of the enormous flexibility in designing and synthesizing aptamers, researchers have functionalized synthetic hydrogels with aptamers having different affinities to PDGF-BB to enable sustained release of the growth factor with a high degree of control over the release kinetics (Figure 14.2) [226,227]. Another special feature of aptamer-based protein conjugation is that binding may be readily reversed by introducing the oligonucleotide sequence complementary to the aptamer. This feature was elegantly utilized to achieve specific and triggered release of multiple growth factors from a microparticle-hydrogel hybrid system [228]. Here, polystyrene microparticles were functionalized with aptamers directed against VEGF and PDGF-BB. The microparticles were then loaded with the growth factors and incorporated into hydrogels. Addition of the specific complementary sequences led to the independent release of the target growth factor without any significant effect on the release of the other growth factor. The high degree of control coupled with the low immunogenicity and toxicity of oligonucleotides makes aptamer-functionalized hydrogels a very attractive targeted drug delivery system.
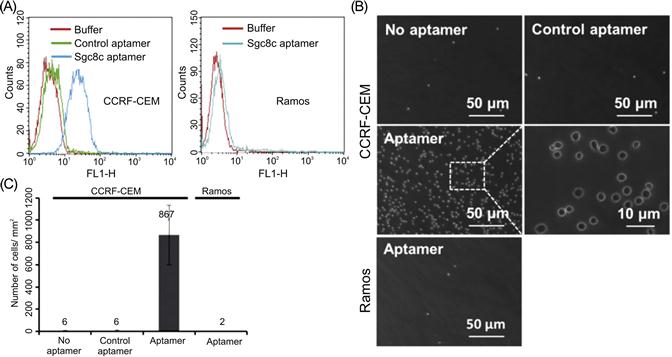
Aptamers can also be selected to engage specific cell types [229]. This is based on cell-specific variations in surface proteins, including receptors and other transmembrane proteins. For example, aptamers that specifically bind to leukemia cells with sub-nanomolar affinity were recently generated and conjugated to PEG hydrogels to facilitate separation and capture of leukemic cells (Figure 14.6) [230]. Adherent cells could also be selectively released under biocompatible conditions by the addition of an endonuclease that cleaved the aptamers [231] or of an oligonucleotide sequence complementary to the aptamer [232].
14.3 Engineering Degradability into Hydrogels
Degradation of hydrogels is essential for many purposes. In tissue engineering applications, degradation is required to create space in the matrix for cell migration and proliferation as well as to allow infiltration of blood vessels. When hydrogels are used for in vivo delivery of drugs, degradation is important for both drug release and removal of the hydrogel scaffold from the body. Importantly, the optimal degradation rate may vary significantly from one application to another, creating the need for tunable degradation kinetics. Moreover, as previously mentioned, the ECM is continuously remodeled by the action of cell-secreted enzymes, so for applications involving tissue regeneration, hydrogels must be amenable to this cell-directed degradation, with the rate of degradation ideally mirroring the rate of new tissue outgrowth. For applications requiring controlled release of therapeutic molecules, the kinetics of degradation should be adequate to maintain effective dosages of the bioactive molecule. A major consideration for hydrogels intended for in vivo implantation is that the degradation products should be nontoxic and amenable to clearance from the body. Major mechanisms of hydrogel degradation that are currently used include hydrolysis and enzymatic degradation.
14.3.1 Hydrolytic Degradation of Hydrogels
Hydrolysis causes homogenous degradation of hydrogels carrying hydrolytically sensitive macromers and crosslinkers. This leads to changes in the overall network properties and can significantly influence the diffusivity of entrapped drugs and proteins [233]. Hydrolytic degradation is spontaneous and is often preferred over enzymatic degradation in drug delivery applications, where patient-to-patient variability in the levels of circulating enzymes can lead to unpredictability in the release of drugs from enzymatically degradable hydrogels [234–236].
Hydrolytically degradable hydrogels most commonly utilize the hydrolysis of ester linkages. This mechanism has been applied for fabrication of both natural and synthetic polymer-based matrices. HA functionalized with hydrolysis-sensitive poly(lactic acid)-methacrylate (PLA-MA) has been crosslinked using the polymerization reaction of methacrylate to fabricate hydrolytically degradable hydrogels that allowed controlled release of VEGF [237]. HA has also been crosslinked with other hydrolyzable crosslinkers such as glycidyl methacrylate [238] and PEGDA [96] in order to make hydrogels susceptible to hydrolysis. Hydrogels based on synthetic polymers such as PEG have also been made sensitive to hydrolysis by incorporating hydrolyzable polymers like poly(lactic acid) (PLA) and poly(glycolic acid) into the scaffolds. The most notable example of such networks has been made using triblock polymers of the form PLA–PEG–PLA with acrylate end groups for crosslinking [81]. The ratio of PEG to PLA segments can be used to manipulate the degradation rate as well as permeability [81,239]. Degradable PEG hydrogels have also been fabricated by reacting an ester containing, amine-reactive PEG derivative with a branched PEG amine [240]. Proteins could be covalently immobilized onto this completely hydrophilic hydrogel via reaction of lysine residues with the amine-reactive groups. Multiarm PEGs terminated with hydrolyzable acrylate moieties have also been crosslinked with a PEG dithiol using the Michael addition reaction [241]. Multiarm PEGs terminated with vinylsulfone have also been often used to form hydrogels [242]. Unlike the acrylate group used previously, the vinylsulfone moiety is not hydrolyzable, and so dithiol crosslinkers containing ester groups have been used in order to render these hydrogels susceptible to hydrolytic degradation. It has been further demonstrated that the hydrolytic degradation rates of such PEG-based synthetic hydrogels can be independently modulated without affecting other physical properties. This was first accomplished by using acrylate-terminated PEG macromers incorporated with ester linkages with variable alkyl chain length (acetyl, propionyl, and butyryl esters) [243]. While the mechanical properties of the three formulations were very similar, the degradation rates were significantly higher for shorter alkyl chain lengths. Hydrolysis rates of PEG-based hydrogels have also been controlled by varying the charge of the cross-linker [244]. In this study, acrylate-terminated multiarm PEG was crosslinked using peptides containing either arginine (positively charged) or aspartate (negatively charged) residues. The arginine-based gels underwent hydrolysis 12 times faster than the aspartate-based gels.
14.3.2 Enzymatic Degradation of Hydrogels
Degradation of hydrogels by cell-secreted enzymes is often desirable in tissue engineering systems where there is an interest in locally restricting degradation to regions of cell invasion and coupling the rate of degradation with the rate of tissue formation.
Hydrogels based on natural polymers are sensitive to several cell-secreted enzymes; for example, collagen is degraded by collagenases MMP-1 and MMP-8 [245]. Therefore, hydrogels combining collagen with synthetic polymers such as PEG are also susceptible to cell-mediated degradation [246]. Fibrin hydrogels can be degraded by the action of cell-secreted MMPs and plasmin [247]. HA can be degraded by hyaluronidases [248] and the degradation rate of HA hydrogels can be controlled by covalently modifying the carboxylic groups of the macromer prior to gelation [249]. Hydrogels based on heparin can be degraded by the action of heparanase [250].
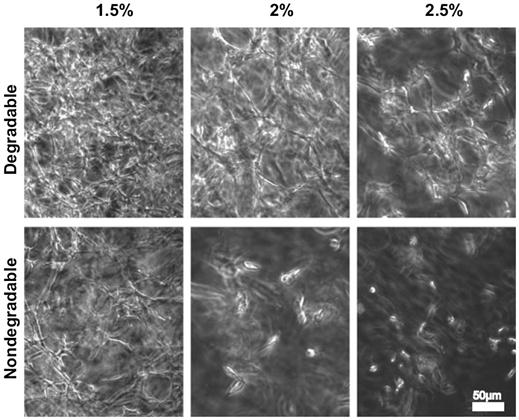
Hydrogels based on synthetic polymers can be made susceptible to enzymatic degradation as well. This has been accomplished by both functionalizing constituent macromers with enzymatic substrates or using crosslinkers consisting of peptide sequences that contain substrates for enzymes such as MMPs. These strategies have been successfully applied for creating degradable hydrogels using PEG molecules incorporating enzymatic substrates at both termini [251]. These hydrogels were crosslinked through photopolymerization of acrylate moieties at the ends of the enzyme substrate peptides, including substrates for the collagenase MMP-1 and plasmin. In another study, inclusion of an elastase-sensitive peptide in a similar model made the hydrogel sensitive to this enzyme [252]. The bioactivity of the hydrogels was further enhanced by including PEG macromers containing the cell-binding peptide KQAGDV into the polymerization reaction. This led to the formation of hydrogels with cell adhesive function and enzymatic degradability. Smooth muscle cells (SMCs), which are known to secrete elastase during migration, could be viably encapsulated in the hydrogels during polymerization. It was demonstrated that the SMCs produced more collagen in the degradable hydrogels than in nondegradable hydrogels. Similar methodologies have also been applied to multiarm PEGs, where the PEG arms were functionalized with collagenase substrates prior to crosslinking [253]. Preadipocytes cultured in these degradable hydrogels formed extensive networks resembling adipose tissue while only isolated cells could be seen in the control nondegradable hydrogels. Furthermore, the intracellular lipid droplets in degradable hydrogels were considerably enlarged, resulting in many unilocular cells, a typical feature of mature adipocytes. Multiarm PEGs with vinylsulfone functionalities have also been crosslinked with MMP-substrate peptides with terminal cysteine residues to fabricate degradable hydrogels [254]. This system allowed precise control over the degradability, matrix elasticity, and the concentration of matrix-conjugated RGD cell adhesive peptides.
MMP-1 substrate peptides have also been combined with the FXIIIa-mediated cross-linking strategy described earlier in order to fabricate MMP-1 and plasmin-sensitive PEG-based hydrogels under highly biocompatible conditions [193,255]. In this strategy, the enzymatically degradable peptide is combined with the FXIIIA substrate peptide (containing the donor FKGG sequence) in order to incorporate degradable motifs into the crosslinks. The ability to independently control biofunctionality, proteolytic degradability and crosslinking density in such hydrogels has allowed the study of the role of matrix stiffness and remodeling on 3D cell migration and formation of cellular networks (Figure 14.7) [255]. This study showed that preosteoblastic cells migrated equally efficiently in soft matrices irrespective of their degradability. However, in stiffer matrices, proteolytic degradation was necessary to facilitate migration. A similar trend was also observed in the ability of the cells to form networks in the hydrogels.
Interestingly, natural polymers have also been crosslinked using peptide sequences containing protease substrates in order to make the hydrogels susceptible to different enzymes. HA has been crosslinked using peptides containing MMP-2 substrate sequences, which rendered the hydrogels degradable by this protease [256]. For this, HA was first modified with both maleimide and methacrylate groups. The maleimides were subsequently reacted with a mixture of RGD peptides with one cysteine residue and MMP-substrate peptides with cysteine residues at each end to enable crosslinking. The methacrylate groups could also be photo-crosslinked to make the hydrogel matrix less sensitive to MMP-mediated degradation. This strategy of having two different crosslinkers allowed independent control over matrix mechanics and degradability. It was demonstrated that human mesenchymal stem cells (hMSCs) which do not secrete hyaluronidase could efficiently degrade the hydrogel by cleaving the MMP-sensitive peptides. Interestingly, the differentiation of these cells was directed by the degradability of the hydrogels independent of the matrix stiffness, with more osteogenic differentiation in the degradable hydrogels. A similar MMP-2 substrate peptide sequence has also been used to crosslink dextran to fabricate cell-degradable hydrogels which are otherwise nonhydrolyzable [257].
14.4 Hydrogel Nanoparticles
Hydrogels can thus be modified with a variety of biomolecules in order to fabricate a biologically instructive microenvironment for cells. Such bioactive hydrogels are used for tissue engineering applications as well as for encapsulating cells for in vivo delivery. Another major area of application of hydrogels has been in the field of controlled delivery of protein drugs such as antibodies and growth factors. Hydrophilic hydrogels provide a biocompatible environment for proteins that can protect them from denaturation, enzymatic degradation, and increase their in vivo circulation time. These properties make hydrogels an attractive medium for engineering the surface of therapeutic cells with exogenous proteins. Synthetic nanoparticles loaded with cytokines have been used to engineer the surface of therapeutic cells in order to increase their in vivo viability and function [258]. Drug-loaded synthetic nanoparticles have also been used for functionalization of cells for targeted delivery using the natural homing behavior of immune cells such as T cells and macrophages [259,260]. Synthetic nanoparticles have also been independently used as delivery vehicles for a variety of therapeutic proteins [261–263]. The following section will describe a few approaches that have been used for the fabrication of hydrogel nanoparticles or nanogels.
14.4.1 Water-in-Oil Heterogeneous Emulsion
Water-in-oil (W/O or inverse emulsion) methods involve the formation of aqueous droplets (micelles) of hydrophilic polymers in a continuous oil phase by using oil-soluble surfactants, followed by crosslinking of the polymers using water-soluble crosslinkers (Figure 14.8). By using a relatively high concentration of the surfactant, micelles in the submicron range can be obtained, which then form nanogels after crosslinking [264]. Inverse emulsion methods have been used to fabricate chitosan nanogels loaded with the antitumor drug doxorubicin coupled with dextran [265]. In this study, a nanogel diameter of ~100 nm was discovered to favor tissue delivery via the enhanced permeability and retention (EPR) effect commonly exploited to deliver contrast agents and drugs to solid tumors. A similar strategy has also been applied for the preparation of HA-based nanogels [266]. Specifically, thiolated-HA was crosslinked in reverse micelles containing small interfering RNA (siRNA) to form nanogels with a diameter of 198 nm in order to enable intracellular delivery of these molecules for gene silencing. Inverse emulsion techniques have been extensively applied for the production of nanogels based on synthetic polymers such as poly(N-isopropylacrylamide) [267,268], polyacrylamide (PAAm) [269,270], poly(dimethylacrylamide) [271], poly(vinylpyrrolidone) [272,273], and the amphiphilic polymer Pluronic® [274]. Inverse emulsion has also been combined with controlled radical polymerization strategies such as atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT) polymerization to produce nanogels with well-controlled size distributions. This approach was used to fabricate stable nanogels of well-controlled water-soluble poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEOMA) with a very narrow size distribution [275,276]. Similar strategies have been used to synthesize nanogels based on PAAm [277] and PEO-b-PHEMA block copolymers [278].
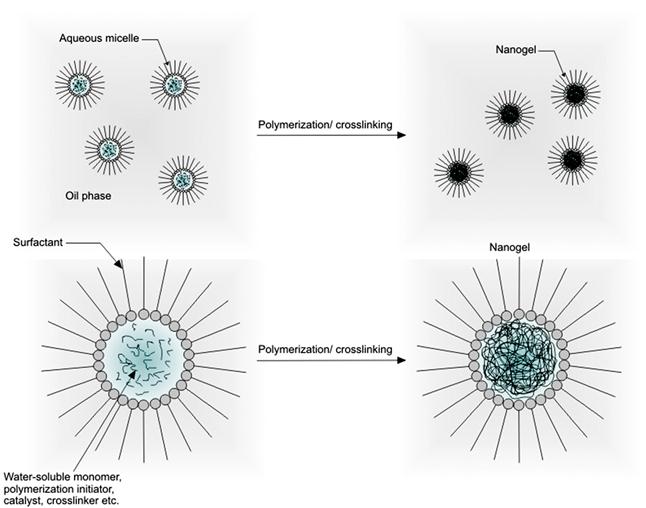
14.4.2 Dispersion/Precipitation Polymerization
In dispersion/precipitation polymerization, the monomer, the polymerization-mediating species and the initiator are initially soluble in water to form a homogeneous solution. As the polymerization proceeds to a critical point following initiation, the generated polymers become insoluble in water and thus form a separate phase. RAFT precipitation/dispersion polymerization of N-isopropylacrylamide using poly(N,N′-dimethylacrylamide) has been used to synthesize core–shell thermoresponsive nanogels [279], which could then be selectively functionalized in the core and the shell using orthogonal chemistry [280]. Aqueous dispersion RAFT polymerization has been used for the synthesis of biocompatible, antifouling, and thermosensitive core–shell nanogels based on copolymerization of oligo(ethylene glycol) methacrylates of different side chain length [281]. A similar strategy based on RAFT-controlled radical crosslinking copolymerization of N,N-diethylacrylamide (DEAAm) and N,N′-methylene bisacrylamide in aqueous dispersion polymerization was also used to synthesize PEGylated thermoresponsive core–shell nanogels [282].
14.5 Conclusion
Hydrogels, with their water-filled network structure and biocompatible nature, are excellent in vitro structural mimics of the cellular microenvironment. However, they often lack the enormous biochemical diversity that is present in the native ECM. In this chapter, we have described traditional and emerging bioconjugation strategies that have been developed to functionalize hydrogels with a variety of biomolecules in order to convert them from inert scaffolds to complex microenvironments that can not only interact with cells but can also be modified by them. This has in turn fueled many successful efforts to integrate growth factors and ECM-derived proteins and peptides to confer important bioactivity to otherwise inert materials. More recently, the ability of specific ECM components (whether native or synthetic) to sequester growth factors and cytokines has been exploited to concentrate cell-derived factors and enhance autocrine and paracrine signaling to promote tissue function. All of these strategies may be aided in the future by new peptide, protein, and aptameric tags that can be fused to target proteins and hydrogels in order to enable site-specific and selective immobilization of factors onto matrices. Moreover, the newest generation of scaffolds has begun to accommodate and direct biodegradability in highly specific ways, most notably by incorporation of crosslinks that may be enzymatically degraded to permit tissue growth and vascularization. Finally, the fabrication of hydrogel nanoparticles with “intelligent” material properties represents an important new frontier in drug delivery systems. A key recurring theme in these newest approaches is the desire to design material properties that optimally interface with biological systems. Whereas traditional biomaterial design may have traditionally focused on choosing a material that “does the least harm” in a given biological system, we are rapidly entering an era in which materials can be designed to both facilitate desirable biological processes and actively direct them.
List of Abbreviations
ADAMTS A disintegrin and metalloproteinase with thrombospondin motifs
BMP Bone morphogenetic protein
CTGF Connective tissue growth factor
IGF Insulin-like growth factor
PDGF Platelet-derived growth factor
PEGDA Poly(ethyleneglycol)-diacrylate
TGF Transforming growth factor